









-
Phalaenopsis is perennial in Orchidaceae, which is one of the four major ornamental orchids (Phalaenopsis spp., Dendrobium spp., Cattleya spp., and Oncidium spp.) in the world[1]. It is named after its special butterfly-like flowers, is gorgeous in color, and has a long flowering period. With the improvement of orchid breeding technology, the Phalaenopsis industry has been rapidly developed, and the number of varieties registered in the Royal Horticultural Society (RHS) has reached nearly 40,000[2−4]. The diverse Phalaenopsis hybrids are highly sought after for their vibrant flower colors, captivating form, and plant morphologies[5].
In recent years, propelled by advancements in high-throughput sequencing technology, several research teams have successfully completed the whole genome sequencing of various Phalaenopsis species, including Phalaenopsis equestris and Phalaenopsis aphrodita[6,7]. These high-quality genome assemblies and annotations have not only elucidated the structural and functional intricacies of the Phalaenopsis orchid genome but have also laid a solid foundation for subsequent gene function research and varietal improvement. Concurrently, the development and implementation of molecular markers have enabled the construction of genetic maps for Phalaenopsis. These genetic maps are pivotal for conducting genetic linkage analysis, quantitative trait locus (QTL) mapping, and marker-assisted breeding. Among the molecular markers, single nucleotide polymorphism (SNP) markers have gradually ascended as the most favored option due to their relatively low cost, high throughput, excellent stability and reproducibility, uniform genomic distribution, and ease of documentation.
SNP markers have been widely used in cultivar recognition, such as Sugarcane[8], Cigar tobacco[9], Brassica napus[10], cucumber[11], wheat[6,12], rice[13,14], cotton[15], and broccoli[16]. Due to the large number of varieties and the small differences in phenotypic traits between varieties, some Phalaenopsis varieties have been identified incorrectly, which has caused great difficulties in the collection, cataloging, preservation, and breeding of new varieties of Phalaenopsis[1,17,18]. Therefore, an efficient, accurate, and economical method is urgently needed to support the breeding work of Phalaenopsis and protect the variety rights. By using SNP fingerprints, breeders can systematically identify and evaluate the germplasm resources and important varieties.
In this study, an SNP database of the Phalaenopsis orchid was developed based on the re-sequencing data to construct the DNA fingerprint map. Through whole genome re-sequencing and variation detection of the Phalaenopsis orchid population, the genetic diversity parameters of Phalaenopsis orchid populations were determined based on SNP data, and the genetic relationship and population structure of Phalaenopsis orchid varieties were further analyzed and confirmed to enrich the genetic variation information. In addition, genetic analysis and variety identification of all germplasm were carried out combined with morphological markers to reveal the rich genetic diversity of Phalaenopsis orchid germplasm. Then the benefits of SNP molecular markers in the study of Phalaenopsis orchid genetic diversity were explored, which provided a reference for the design of molecular marker primers of Phalaenopsis orchid in the future.
-
Fifty-three Phalaenopsis accessions were obtained through collaboration between the Straits Orchid Conservation Center of Fujian Agriculture and Forestry University and Zhangzhou Jubao Biotechnology Company. These accessions were subsequently cultivated in the germplasm resource nursery at the Straits Orchid Conservation Center of Fujian Agriculture and Forestry University (Supplementary Tables S1 & S2), the relevant varieties are used for scientific research purposes and are supported by relevant policies and enterprises. Cultivars were identified by experts in our local lab to obtain accurate characterizations. Young leaves from all samples were carefully selected, promptly frozen using liquid nitrogen, and then preserved in a −80 °C refrigerator to facilitate genomic DNA extraction[19].
Methods for selection and determination of phenotypic traits
-
Considering the ornamental characteristics of Phalaenopsis spp., a study was conducted on 34 phenotypic traits related to leaf, peduncle, and floral organs. Each trait was measured in triplicate to ensure accuracy, and mean values and coefficients of variation (CV) were calculated. The investigation utilized the Royal Garden Society plant colorimetric card, Vernier calipers, steel tape measures, and rulers, identifying 19 quantitative traits and 15 qualitative traits. Phenotypic traits of Phalaenopsis were assessed during the flowering stage, with three biological replicates established. Flower-types are categorized based on their diameters: large flowers exceed 10 cm, medium flowers range from 6 to 10 cm, and small flowers measure less than 6 cm in diameter. For quantitative traits, measurements were averaged, while qualitative traits were described, counted using population observation methods, and assigned corresponding values (Supplementary Table S3).
Data processing of phenotypic traits
-
The data were processed to calculate the coefficient of variation for each trait. Factor analysis was then applied to perform the primary classification of the 34 traits[20]. Finally, SPSS 26 was used to conduct Q-type cluster analysis of Phalaenopsis varieties.
DNA extraction
-
The sample leaves were mashed with liquid nitrogen in a mortar. DNA was extracted by the modified cetyltrimethyl ammonium bromide (CTAB) method, and DNA concentration, integrity and purity were detected by agarose gel electrophoresis, Qubit Fluorometer and enzyme labelometer[21].
Genome resequencing and SNP marker screen
-
The qualified DNA was randomly fragmented using a Covaris ultrasonic disruptor. Large DNA fragments were enriched and purified with magnetic beads, followed by end repair and A-tailing. Circular sequencing adapters were then ligated to both ends of the DNA fragments, and unligated fragments were removed with exonuclease to construct a DNA library. After PCR amplification and library quality assessment, sequencing was performed on the DNBSEQ-T7 platform. The resulting primary image data were converted into raw data. Sequencing adaptors and low-quality reads were filtered out using fastp (v0.23.2), which excluded low-quality reads, reads containing adapters or primer sequences, and reads where either single-end read contained more than 10% 'N' bases or more than 20% low-quality bases (Q ≤ 5); in such cases, the entire paired read was discarded[22]. FastQC (v0.11.8) was then used for quality control, analyzing GC content, base quality, and sequence duplication levels in the filtered data. The final high-quality reads were retained for downstream analyses.
The filtered reads were aligned to the modified reference genome of Phalaenopsis aphrodita, downloaded from the Orchidstra2 database (
https://orchidstra2.abrc.sinica.edu.tw/orchidstra2/index.php ) using BWA software (v.0.7.17) with default settings[23,24]. Duplicate reads were marked and removed using the GATK MarkDuplicates function (v4.0.10.0)[25]. SNPs were filtered using a sliding window approach, removing SNPs where five or more appeared within a 10 bp window. Additional quality filtering criteria for SNPs included: QD < 2.0, MQ < 40.0, FS > 60.0, SOR > 3.0, MQRankSum < −12.5, or ReadPosRankSum < −8.0[26]. The filtered SNPs were annotated using SnpEff (v5.1). Further filtering of the VCF files excluded loci with a missing data rate greater than 50% and those with a minor allele frequency (MAF) below 0.1; the remaining loci were used for genetic diversity analysis[27]. Additionally, VCFtools was used to analyze SNP markers, calculating metrics such as Minor Allele Frequency (MAF), Polymorphism Information Content (PIC), and Nucleotide Diversity Index (Pi).Analysis of genetic diversity
-
For reference, plink (v1.90b6.21) was utilized for LD filtering of SNPs data, removal of SNPs with a deletion rate exceeding 10% minor allele frequency impacting accuracy in population samples, and retention of secondary loci[28]. Subsequently, plink was employed for SNP filtering (with individuals within each subpopulation required to adhere to Hardy-Weinberg equilibrium and each SNP site being independent and unlinked)[29]. The processed data was then filtered using plink for PCA analysis. Population PCA analysis scatter plots were generated using the ggplot2 package in R (
www.r-project.org ). For population structure analysis, admixture software (v1.3.0) was used to define multiple population subgroups or ancestral numbers (K = 1~15), calculate error rates of sample coefficient variation under different K values, visualize results in R, and generate distribution maps of CV errors. The optimal K value is determined as the one with the lowest cross-validation error rate, and genetic structure mapping is conducted using the ggplot2 package in R under this K value setting. The format of SNPs data was converted using phylip (v3.697) (converting .vcf files into .fa files), followed by comparison using MEGA11 and construction of a phylogenetic tree utilizing the NJ method[30], with Bootstrap testing support rates for each branch set at 1,000 repeats. Finally, ITOL (https://itol.embl.de/ ) was utilized to enhance the visual presentation of the tree structure[31].DNA fingerprinting
-
The 53 Phalaenopsis germplasm were genotyped using the SNP loci obtained from the screening and DNA fingerprinting was performed based on the nucleotide sequence information. Each variety was then encoded with a unique barcode generated by an online software (
http://qr-batch.com/ ). The variety name and fingerprint code information of each germplasm were input to generate a two-dimensional code for each variety. -
Genetic diversity analysis was conducted on 19 quantitative and 15 qualitative traits across 53 Phalaenopsis cultivars (Fig. 1b, c). The CV for quantitative traits ranged from 16.09% to 154.60%, with an average of 37.35%, indicating substantial phenotypic variability among cultivars. Notably, traits with the highest CVs—such as the number of lateral branches on the flower stalk and the number of flowers per inflorescence—highlight significant morphological diversity, although these traits alone do not fully support a conclusion of low trait stability across all cultivars. For qualitative traits, the CV ranged from 20.54% to 130.81%, averaging 50.59%, which reflects considerable variability in quality traits, particularly for characteristics like sepal margin waviness and flower fragrance. Together, the broad CV ranges observed in both quantitative and qualitative traits suggest high morphological diversity.
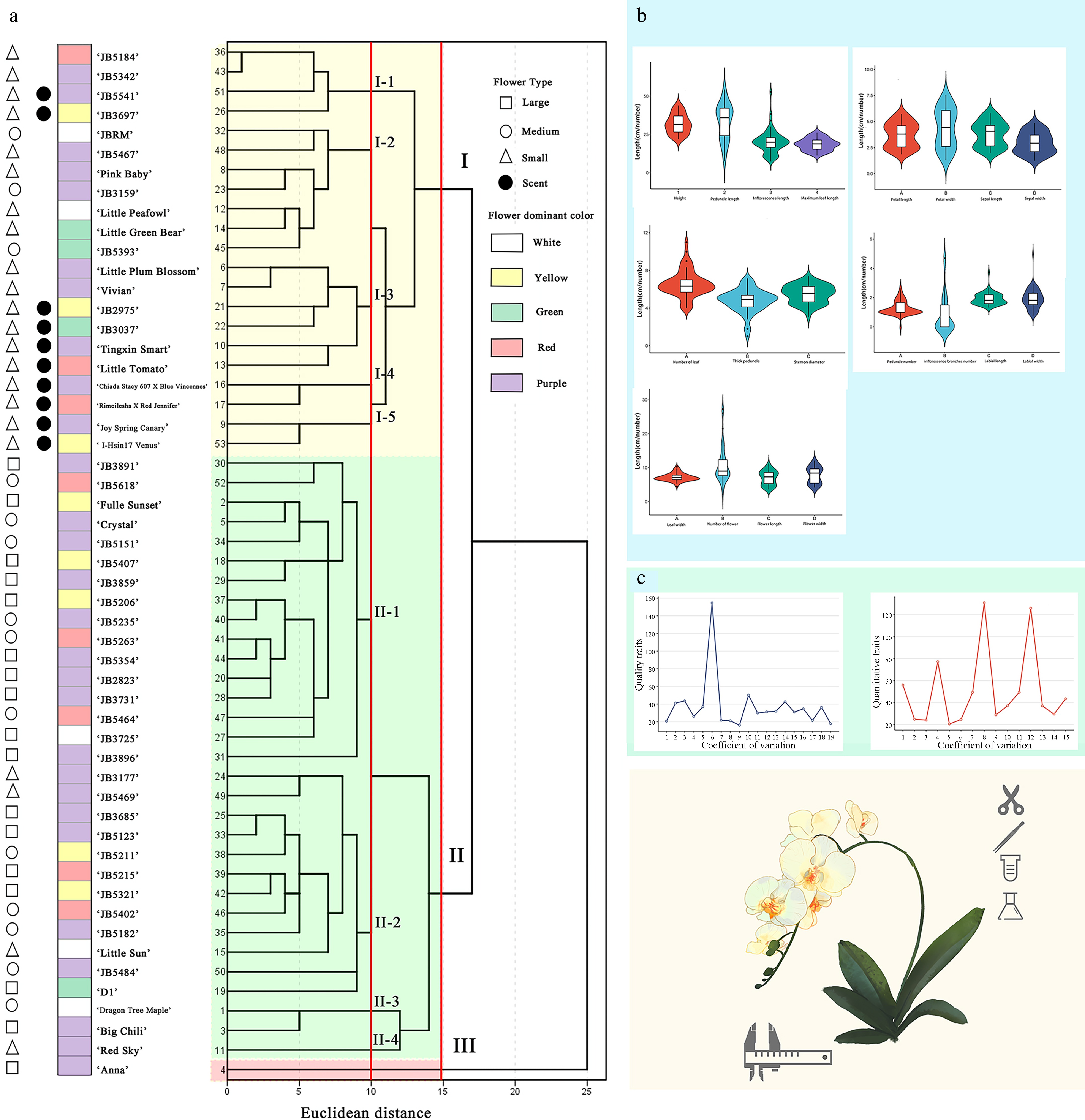
Figure 1.
(a) Q-type cluster analysis of the 53 Phalaenopsis cultivars revealed genetic distances ranging from 1 to 25; (b) The mean value of 19 quantitative traits; (c) Coefficient of variation of 19 quantitative traits and 15 quality traits. (For specific trait characteristics, see Supplementary Table S2).
A Q-type cluster analysis of the 53 Phalaenopsis cultivars revealed genetic distances ranging from 1 to 25 (Fig. 1a). At a Euclidean distance of 15, the cultivars were grouped into three main clusters, demonstrating significant genetic diversity within the tested Phalaenopsis collection. Group I comprised 21 cultivars, Group II included 31 cultivars, and Group III contained only the 'Anna' cultivar. Notably, none of the other 52 cultivars were the direct progeny of 'Anna'. The results suggested that greater phenotypic and genetic differences among parent cultivars yield more genetically diverse progeny, a desirable trait for cultivar improvement. Additionally, Group I predominantly comprised small and medium-flowered types, while subgroups II-1 and II-3 included large and medium-flowered types[32]. Subgroups I-4 and I-5 contained fragrant-flowered cultivars. Interestingly, large and small-flowered cultivars rarely appeared in the same subgroup, indicating a correlation between flower size and clustering. Fragrant cultivars were all found in subgroup I, suggesting a potential link between fragrance and cluster branching. However, flower color was not strongly correlated with cluster structure, as multiple colors (e.g., reddish-purple, red, yellow, white, and green) were present within subgroups I and II.
Significance analysis of the 19 quantitative traits by flower type revealed that traits associated with floral organs exhibited strong significance, indicating that the main differences among the tested Phalaenopsis are concentrated in floral morphology, with weaker correlations to leaf traits (Supplementary Fig. S1). Based on the Q-type clustering results at a Euclidean distance of 15, a principal component analysis (PCA) was performed on the 53 samples divided into three groups (Fig. 2a). The first two principal components accounted for 38.4% and 8.1% of the total genetic variance, respectively. Samples exhibited significant inter-group differences on PC1, while PC2 contributed less. The confidence ellipses of groups A and B were distinctly separated, indicating clear group characteristics. In PC1, traits with high information loadings were flower length (0.938), flower width (0.978), petal length (0.971), petal width (0.964), sepal length (0.956), and sepal width (0.935), suggesting that the first principal component is primarily associated with floral organ traits. In PC2, traits with high loadings were leaf tip symmetry (0.622), leaf surface type (0.524), and flower surface waxiness (0.702), indicating that leaf-related traits played a major role. PCA by flower type showed that the confidence ellipses of groups A and C had almost no overlap, demonstrating significant differences in the principal component characteristics between these two groups (Fig. 2b). The confidence ellipse of group B partially overlapped with those of groups A and C, indicating a greater diversity in the principal component characteristics of group B, suggesting that medium-flowered Phalaenopsis may have undergone more extensive genetic exchange.
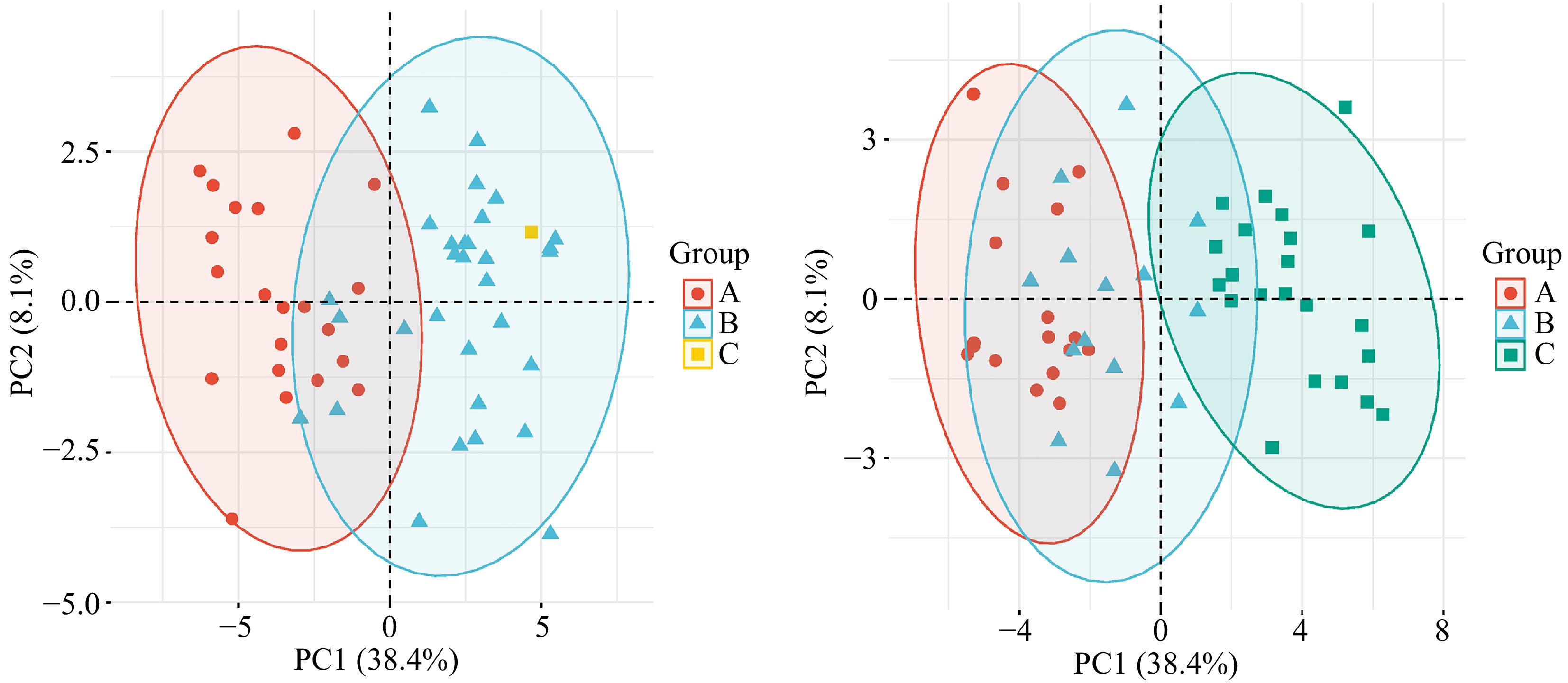
Figure 2.
Based on 34 phenotypic traits, PCA analysis of 53 species of Phalaenopsis.
Mapping to the reference genome and filtration of SNPs
-
These high-quality SNPs were uniformly distributed across the Phalaenopsis genome, except in the centromere regions (Fig. 3a). The statistical analysis of variant types identified 12 distinct types, with C/T (23.86%), G/A (22.31%), A/G (8.89%), and T/C (7.84%) being the most prevalent. The transition to transversion ratio was approximately 1.70. Furthermore, the analysis revealed that 70.91% of the SNP loci are situated in intergenic regions, 9.61% in upstream regions, and 9.37% within intronic regions. Notably, only 1.31% of the SNP loci are located in exonic regions, with a mere 0.03% in splicing regions.
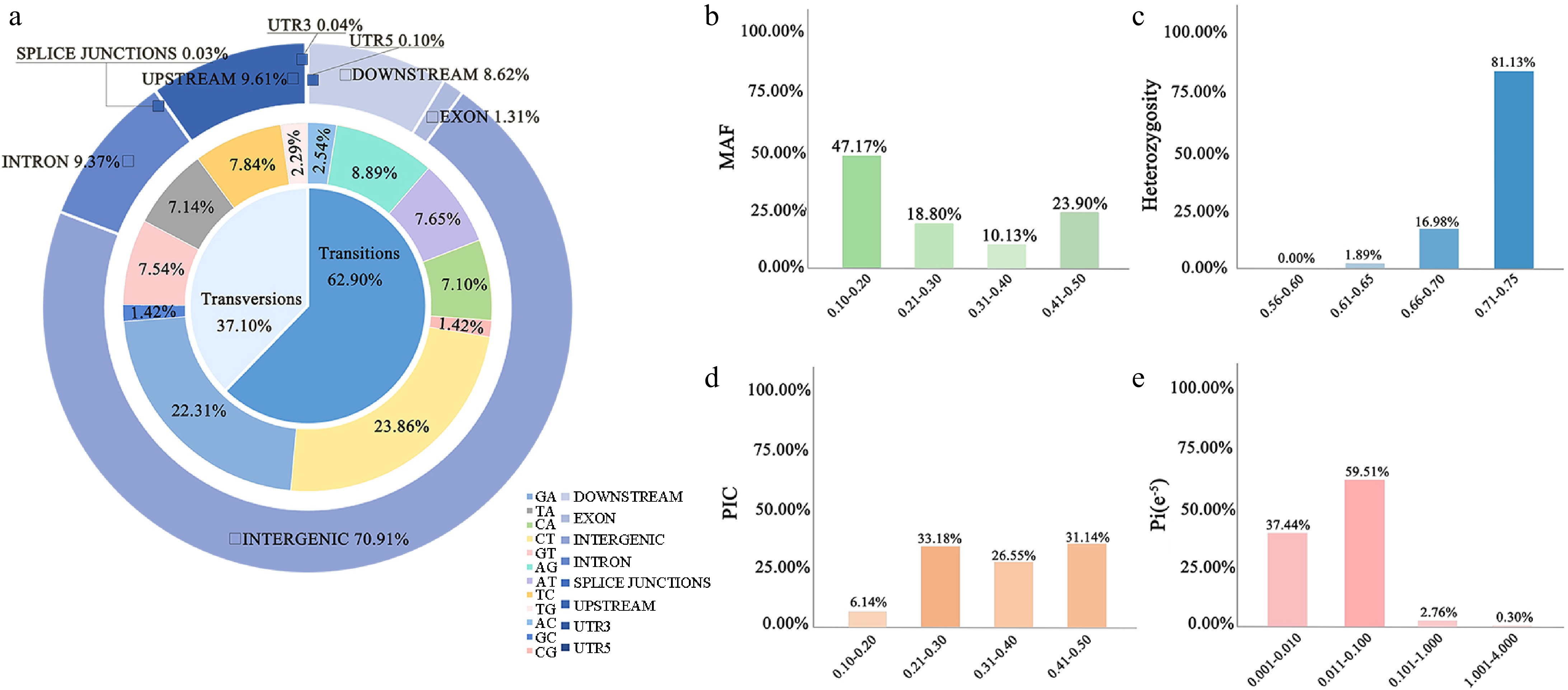
Figure 3.
SNP screening and statistics. (a) The positions of the SNPs in the gene structures. Genotype statistics for the six most commonly identified variants. Genetic information content of 5,984 high-quality markers in 53 cultivars including (b) MAF, (c) heterozygosity rate, (d) PIC, and (e) nucleotide diversity.
We assessed the quality of the 5,984 SNPs by calculating their PIC, MAF, heterozygosity, and nucleotide diversity. The MAF values ranged from 0.105 to 0.500, with an average of 0.272. The PIC values ranged from 0.188 to 0.500, averaging 0.355. Heterozygosity had a mean value of 0.714, ranging from 0.645 to 0.740. Nucleotide diversity averaged 1.103e-6, ranging from 8.99e-9 to 4.70e-5 (Fig. 3). These results indicate that the 5,984 SNPs exhibit high polymorphism and are suitable for DNA fingerprinting analysis in Phalaenopsis.
Analysis of genetic diversity
-
In this study, we conducted an in-depth analysis of the population structure of 53 Phalaenopsis cultivars from diverse geographical regions using Bayesian analysis[33] . The effective population size was estimated through cross-validation of the coefficient of variation (CV error), identifying the lowest CV error at K = 2 (Fig. 4a), which resulted in the samples being subdivided into two distinct categories. Our genetic analysis revealed consistently low mutation rates across K values from 2 to 15 (Fig. 4b). When K = 3, further subdivisions emerged, with genetic components beginning to intermingle, suggesting notable intra-population genetic diversity. As K increased beyond 3, the genetic structure became increasingly complex, likely reflecting contributions from multiple ancestral sources.
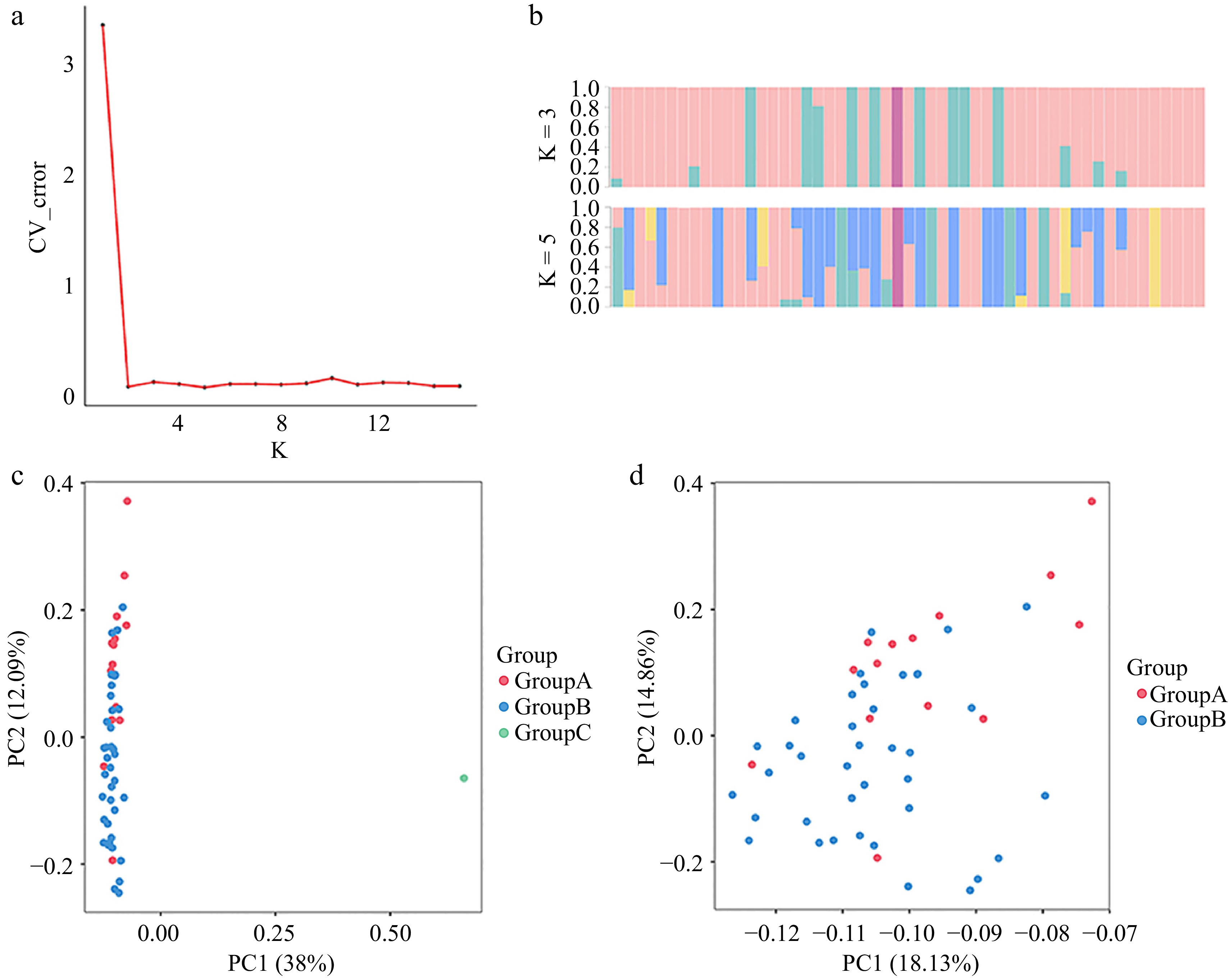
Figure 4.
Genetic diversity analysis. (a) DK values corresponding to different K measurements. (b) Population structure of the 53 germplasm resources at different values of K. (c) Principal component analysis of 53 Phalaenopsis materials. (d) Principal component analysis of 52 Phalaenopsis materials.
Additionally, referring to SNP markers, we conducted Principal Component Analysis (PCA). The optimal K value, determined by the inflection point where the error sharply declined and stabilized, was identified around K = 3. This value was selected as the basis for grouping in PCA. Our analysis categorized the 53 materials into three distinct populations: Group A, Group B, and Group C. Principal components PC1 and PC2 explained 38% and 12.09% of the total genetic variance, respectively. Visualization of the PCA two-dimensional plot illustrated 'JBRM' as an outlier distinctly separate from the main cluster (Fig. 4c). Upon removing 'JBRM', Group A and Group B showed minimal genetic differentiation, suggesting some degree of genetic similarity between these two groups.
To provide a clearer illustration of the genetic relationships among these cultivars, a phylogenetic tree constructed from the 5,984 SNPs offers valuable insights into their evolutionary connections and genetic distinctiveness. Although some branches displayed low support values, the main branches achieved a support rate of approximately 70%, underscoring reliable grouping among the major lineages (Fig. 5). This indicates that SNP data can be effectively utilized to elucidate the genetic relationships within the Phalaenopsis germplasm. The genetic structure divided 53 varieties into five distinct groups, with only one variety present in each of groups II, III, IV, and V. Group I included a larger number of germplasms and was further subdivided into 15 subgroups. Notably, subgroup I-1 contained seven germplasms such as 'Tingxin Smart' and 'Little Tomato', which exhibited fragrant flower characteristics, suggesting an association between the genetic makeup of Phalaenopsis orchids and floral fragrance. Similarly, subgroup I-7 consisted of five germplasms, including 'JB3697', characterized by small flowers, while subgroup I-14 comprised three germplasms, including 'JB3685', with large flowers, indicating a correlation between the genetic relationships of butterfly orchids and flower type.
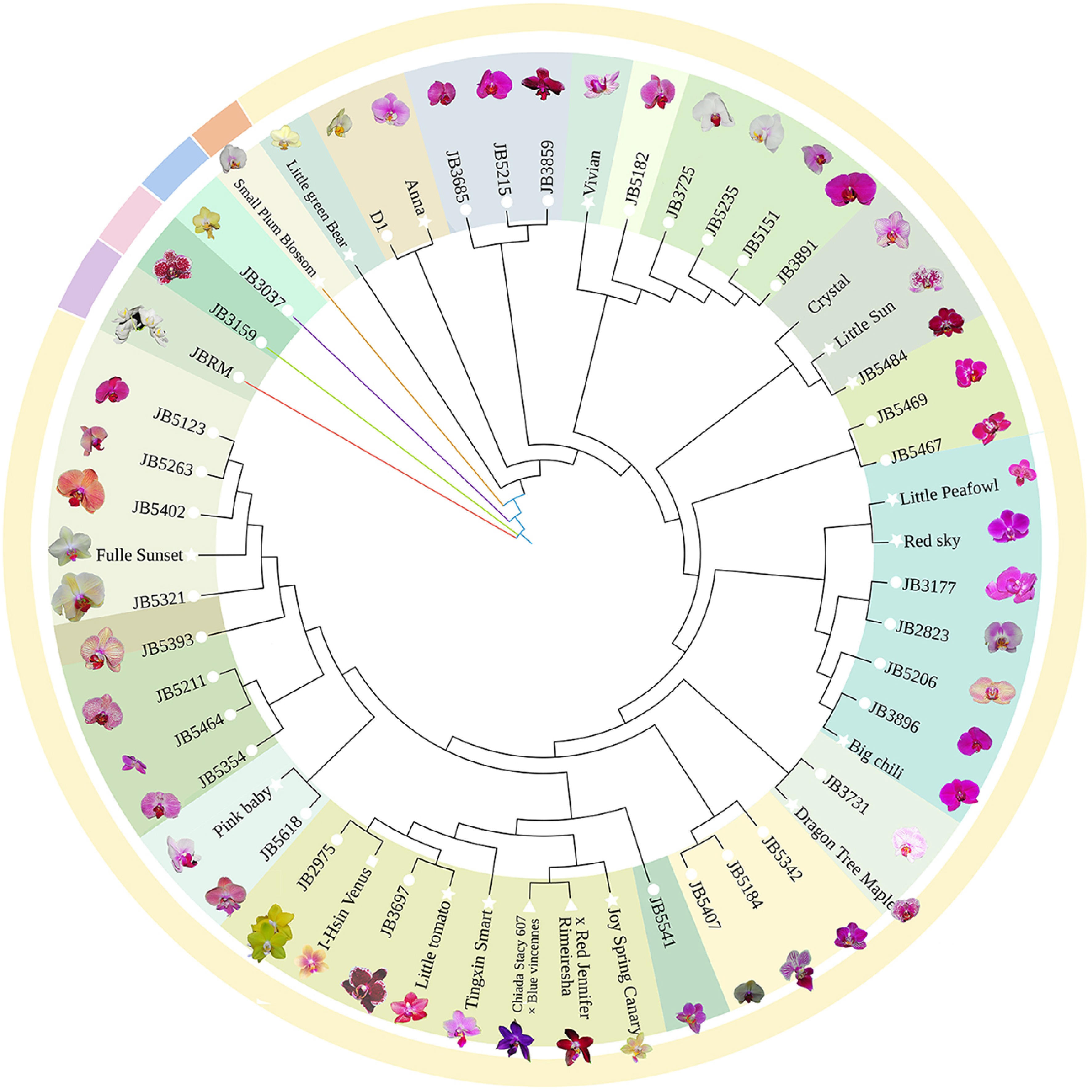
Figure 5.
Phylogenetic tree of 53 cultivars constructed with 5,984 SNP markers.
It is important to note that there was a weaker correlation among homologous flower clusters compared to fragrant flower types; many homologous germplasms exhibited cross-clustering. Additionally, the examination of the evolutionary tree revealed both closely related and distant germplasms within the samples from Zhangzhou. Germplasms from the same source were not entirely clustered together but were instead distributed across different branches. These findings align with the results obtained from principal component analysis, which suggest potential gene exchange among Phalaenopsis varieties.
Screening of core SNP sites and construction of SNP fingerprints
-
Based on 5,984 high-quality SNP loci, we conducted a secondary screening with the parameters (Fig. 6a): MAF 0.1, max-missing 0.8, min-meanDP 3, and hwe 0.01. This process resulted in the identification of 14 core SNP loci, which were evenly distributed across the genome and demonstrated good representation. An NJ tree was constructed based on these 14 core SNP sites (Fig. 6b), and genetic diversity metrics such as PIC values were calculated (Table 1). The NJ tree constructed from the 14 core SNP loci and the tree based on the full set of 5,984 genome-wide SNP loci revealed consistent clustering results among the 53 Phalaenopsis cultivars, highlighting the efficacy of the core loci in capturing essential genetic structure. The tree from 5,984 SNP loci, divided the cultivars into five groups, with Group I further subdivided into 15 subgroups. This detailed subdivision allowed for associations between specific genetic clusters and phenotypic traits, such as fragrance in subgroup I-1 and flower size in subgroups I-7 and I-14. In contrast, the NJ tree based on the 14 core SNP loci produced a comparable but slightly broader clustering, dividing the cultivars into four main populations. Despite the reduced SNP count, the core loci tree preserved key genetic relationships and diversity metrics, with PIC values closely aligned with those obtained from the full SNP dataset. This similarity indicates that the 14 core SNP loci provide a representative framework for distinguishing between the Phalaenopsis cultivars, thus offering a reliable and efficient alternative for genetic analysis in this genus.
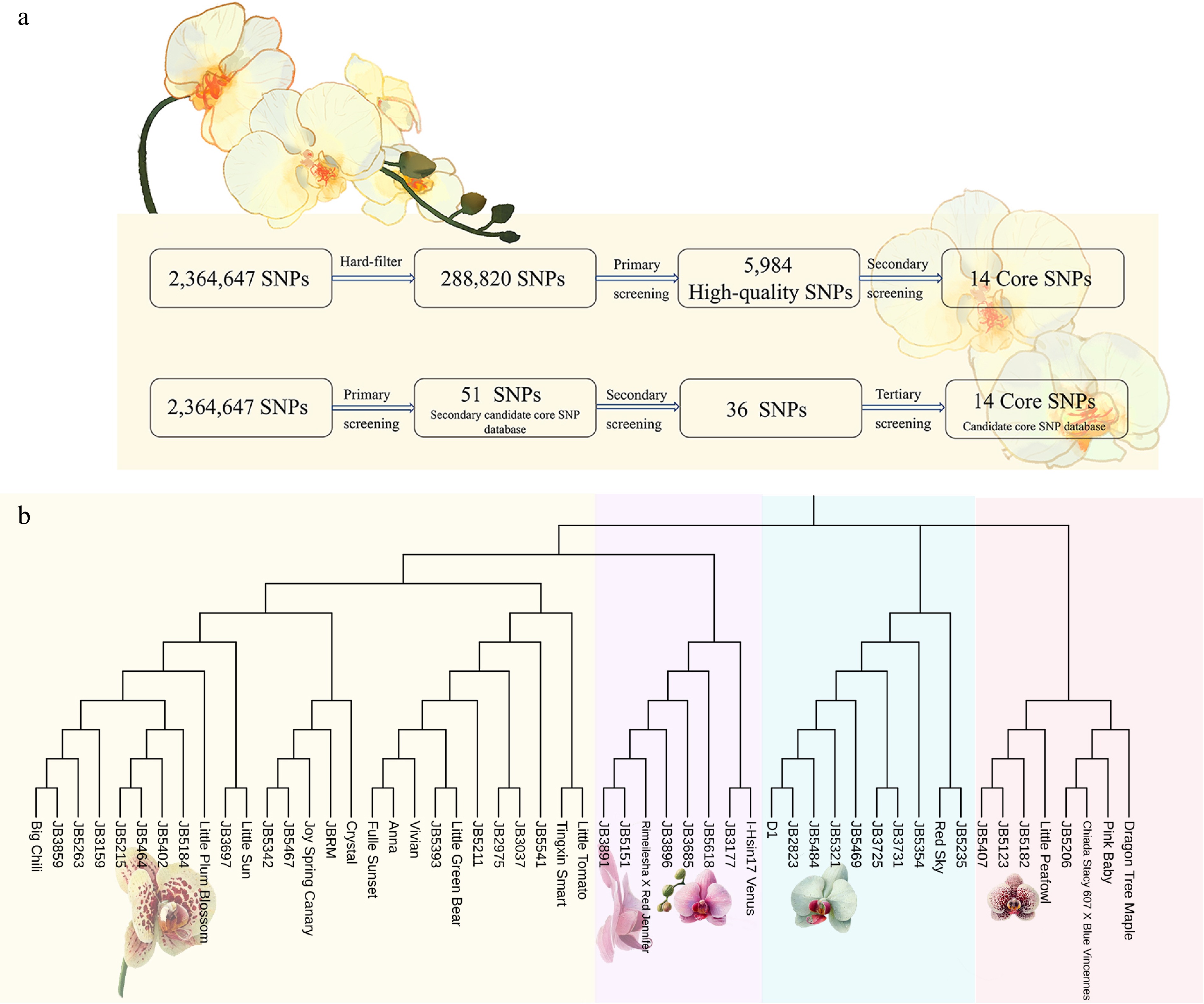
Figure 6.
(a) Flow chart of candidate SNP site screening. (b) NJ tree was constructed based on 14 core SNP sites.
Table 1. Genetic diversity parameter of core SNPs.
Chr Variation type MAF PIC Pi scaffold3 T/C 0.18 0.29 1.36E-04 scaffold17 C/T 0.16 0.27 1.33E-04 scaffold87 T/A 0.12 0.21 9.27E-05 scaffold98 C/A 0.11 0.19 9.19E-05 scaffold138 C/A 0.35 0.45 2.29E-04 scaffold142 G/T 0.19 0.31 1.23E-04 scaffold214 G/T 0.27 0.40 1.86E-04 scaffold260 A/G 0.24 0.36 1.68E-04 scaffold480 A/C 0.15 0.26 1.27E-04 scaffold480 T/A 0.50 0.50 3.01E-04 scaffold562 G/T 0.12 0.20 9.92E-05 scaffold943 C/G 0.28 0.40 1.81E-04 scaffold1243 A/G 0.13 0.22 1.04E-04 scaffold4495 T/A 0.23 0.35 1.77E-04 We propose that these 14 markers serve as suitable core selections for constructing distinct genetic fingerprints for the identified 53 Phalaenopsis varieties (Supplementary Fig. S2). For the analysis, genotypes identified by the aforementioned core markers were converted into numerical codes (Fig. 7): pure genotypes AA = 1 = yellow, CC = 2 = green, GG = 3 = blue, and TT = 4 = pink; heterozygous genotypes were coded as '5' representing orange; and no call genotypes were represented by NA = 6 = white. Subsequently, fingerprinting was conducted using R software, where individual rows represented specific samples and columns represented SNP marker data (Supplementary Table S4).
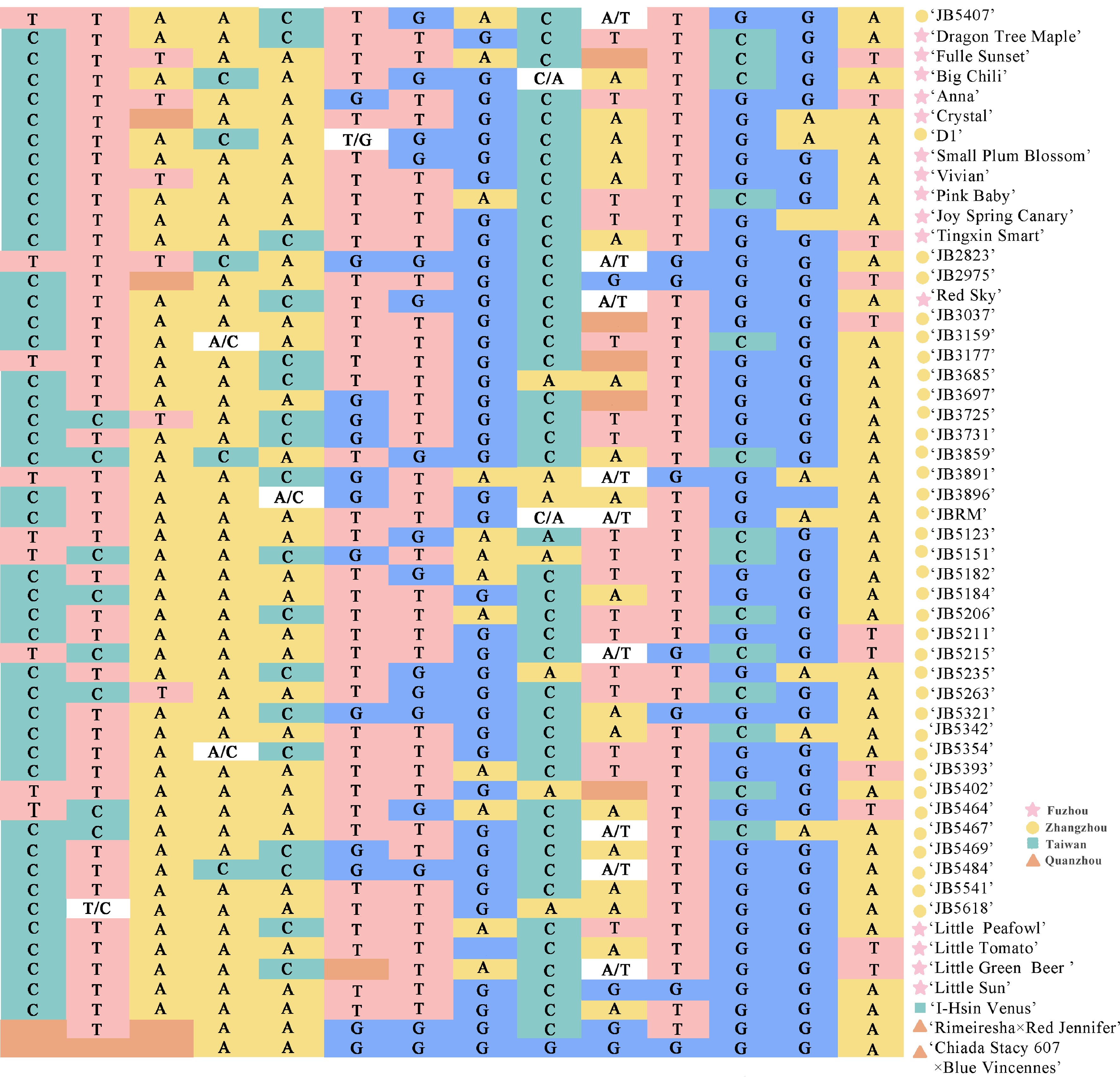
Figure 7.
Fingerprint analysis of 53 Phalaenopsis varieties. Each row represents a sample and each column represents a genome. Pure genotypes are AA = yellow, CC = green, GG = blue, TT = pink; heterozygous genotypes are orange; no call genotypes are white.
-
In previous studies, the identification and genetic diversity of Phalaenopsis species primarily relied on first- or second-generation molecular marker technologies such as RAPD, SSR, SRAP, and ISSR. However, the limited number of genetic marker sites obtained or used constrained the comprehensive exploration of polymorphic sites and genetic information at the whole-genome level. In contrast, SNP markers, as third-generation molecular markers, offer advantages including high stability, precision, an abundance of sites, and direct relevance to phenotypic traits, surpassing other types of molecular markers.
Based on our research, we selected 53 popular market varieties for whole-genome resequencing to efficiently and accurately obtain genetic information and polymorphism loci of Phalaenopsis species. The effectiveness of phenotypic association analysis depends on large scale, high-quality phenotypic data, and highly diverse population backgrounds[34]. In addition, in the specific phenotypic characteristics of Phalaenopsis, the performance of floral fragrances, flower patterns and other traits are usually affected by complex genetic regulation and environmental factors, and large-scale population and environmental repeated experiments are needed to reduce errors caused by environmental changes[35,36]. Therefore phenotypic association analysis was not employed. With the limited sample population size, core SNP screening was conducted based on the screening parameters suggested in previous literature. Although phenotypic association analysis can provide more targeted character-specific SNPs, core SNP screening methods based on the previous research can still generate representative sets of SNPs for germplasm resource identification and genetic diversity analysis under resource constraints. Furthermore, the preliminary results of this study show that the NJ tree of 14 core SNPs is highly consistent with the clustering results based on 5,984 genome-wide SNPs, further demonstrating the validity and representatives of the set of SNPs produced by these screening parameters in reflecting population structure and genetic diversity. Also, in the future, it is possible to combine large-scale phenotypic data to further apply character-based phenotypic-genotype association analysis methods to identify SNPs associated with specific traits (such as flower type, and aroma) and create more accurate fingerprints for each variety. This approach will be more helpful in digging deeper into the genetic basis of the specific phenotype of Phalaenopsis.
Most of the SNP variation sites were located in intergenic regions, with a certain degree of distribution observed in intronic, upstream, and downstream regions, likely due to close interspecific hybridization and frequent gene exchange. This hypothesis was supported by statistical analysis and subsequent principal component analysis (PCA) of phenotypic traits, as well as the construction of evolutionary trees. PCA indicated gene exchange, while genetic structure analysis based on Admixture revealed no significant population structure within the tested Phalaenopsis population. The discovery that core SNPs are distributed across multiple scaffolds may be attributed to the incomplete chromosome-level assembly of the P. aphrodite genome. During sequencing, the genome is typically fragmented into contigs or scaffolds, which are then aligned to form larger assemblies. If gaps exist within the assembly, scaffolds may remain unanchored to specific chromosomes. Consequently, in the absence of chromosome-level resolution, SNPs may be dispersed across various scaffolds rather than being accurately localized to distinct chromosomal regions[36]. At the same time, the loss of centromere regions may also be due to the lack of accurate labeling of these regions in the reference genome itself, which affects mutation detection and SNP localization, resulting in a lack of detailed centromere information in the final genome[37].
The NJ evolutionary tree was established based on 5,984 SNP loci. Clustering divided the 53 germplasms into 19 subgroups, with several relatively independent clusters. Studies suggest that asexual breeding and artificial selection may lead to an accumulation of genetic variation in germplasm, increasing genetic distance, and forming multiple relatively independent clusters. Tissue culture, the primary breeding method in the Phalaenopsis industry, is asexual and follows a stable breeding strategy. It can be inferred that this unique reproduction mode is a significant reason for the indistinct population structure observed. The genetic structure analysis from this study showed that most Phalaenopsis germplasms were aggregated, except for some from Zhangzhou, which were distant from the main cluster. Therefore, these highly heterozygous Phalaenopsis varieties exhibit common inheritance patterns, suggesting complex multiple crosses among their parents. Although this study is limited by sample size, the results accurately reflect the genetic background of Phalaenopsis varieties to some extent and hold substantial reference value.
-
In this study, genome skimming was employed to develop SNP markers. From 2,364,647 potential sites, 5,984 high-quality SNP sites were identified, leading to the establishment of a high-quality SNP database for Phalaenopsis cultivars. Among these, 14 core SNPs were selected for generating related DNA fingerprint codes for 53 Phalaenopsis cultivars various commercial varieties available in the market. The construction of Phalaenopsis DNA fingerprints provides technical support and promotes the development of molecular plant breeding for variety identification, relatedness delineation, and the collection and conservation of germplasm resources.
This work was supported by the National Key Research and Development Program of China (2023YFD1600504), Fujian Provincial Natural Science Foundation of China (2023J01283), and the Key Research and Development Program of Ningxia Hui Autonomous Region (2022BBF02041).
-
The authors confirm contribution to the paper as follows: study conception and methodology: Peng D, Zhou Y, Zhao K; data curation, writing-original draft preparation: Chen X, Wang Q; writing-reviewing and editing: Wang F, Wu X, Guan Y; software: Chen X, Pan Y; formatting, correction: Xue L, Duan Y; charting: Chen X, Wang S. All authors reviewed the results and approved the final version of the manuscript.
-
The raw resequencing data have been deposited in Genome Sequence Archive of National Genomics Data Center under the following accession: CRA020415. All data generated or analyzed during this study are included in this published article and its supplementary information files, and also available from the corresponding author on reasonable request.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 Main information of 53 materials.
- Supplementary Table S2 Phenotypic genetic diversity of 53 Phalaenopsis cultivars.
- Supplementary Table S3 Indicators and criteria of phenotypic diversity of Phalaenopsis.
- Supplementary Table S4 Information of Core SNPs.
- Supplementary Fig. S1 The significance of 19 quantitative characters was analyzed according to flower type classification.
- Supplementary Fig. S2 Phalaenopsis fingerprint two-dimensional code.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Chen X, Wang Q, Wang F, Wu X, Pan Y, et al. 2025. SNP fingerprint database and makers screening for current Phalaenopsis cultivars. Ornamental Plant Research 5: e011 doi: 10.48130/opr-0025-0005 

SNP fingerprint database and makers screening for current Phalaenopsis cultivars
- Received: 10 September 2024
- Revised: 28 December 2024
- Accepted: 03 January 2025
- Published online: 12 March 2025
Abstract: The Moth orchid is globally recognized as one of the most popular and important ornamental species. However, due to the complicated history of hybridization, long growth cycles, and industrial vegetative propagation, there are huge challenges in cultivar identification and protection leading to market issues. Consequently, it is important to develop effective and stable markers to identify and preserve core Phalaenopsis cultivar resources. In this study, we collected 53 commercially prevalent Phalaenopsis cultivars in China. Through detailed phenotypic observations, morphological genetic diversity was measured in 19 quantitative and 15 qualitative traits. By genome skimming and the subsequent SNP calling pipeline, we discovered 5,984 high-quality single nucleotide polymorphisms (SNPs) and constructed a comprehensive Phalaenopsis SNP database of cultivars. These SNPs got a high correlation with variation for quantitative traits ranging from 16.09% to 154.60%, while those for qualitative traits spanned from 20.54% to 130.81%. This database demonstrated a high degree of genetic diversity and a robust capacity for identifying polymorphisms and distinguishing among current varieties. These discovered SNPs consist of 12 types, C/T (23.86%), G/A (22.31%), A/G (8.89%), and T/C (7.84%). The ratio of transition to transversion was approximately 1.70. 70.91% of the SNP loci were in intergenic regions, 9.61% in upstream regions, and 9.37% within intronic regions. Fifty three cultivar PCAs could serve as three groups, which was matched with trait clusters. Based on 5,984 SNP sites, we conducted secondary screening and screened out 14 core sites. The clustering results of the NJ tree based on 14 core SNP loci and the NJ tree based on 5,984 whole genome SNP loci were consistent among 53 Phalaenopsis cultivars. Besides, each variety was then encoded with a unique barcode by 14 core SNP markers. This preliminary approach offers a putative and effective tool for variety identification, genetic analysis, and further development of Phalaenopsis germplasm resources.
-
Key words:
- Phalaenopsis /
- SNP marker /
- Genetic diversity /
- Fingerprinting