










-
MicroRNAs (miRNAs) are endogenous non-coding RNAs, 20−24 nucleotides in length, which play a crucial role in regulating gene expression. They are pivotal modulators of developmental processes and responses to environmental stimuli in eukaryotes. The biogenesis of plant miRNAs consists of three major steps[1,2]. Endogenous MIRNA (MIR) genes are transcribed as independent units by Pol II, generating stem-loop structured pri-miRNAs. These pri-miRNAs are then processed into a miRNA/miRNA* duplex with 2-nucleotide overhangs at the 3' ends by the dicing complex, which consists of the nuclear endonuclease DICER-LIKE 1 (DCL1), the double-stranded RNA-binding protein HYPONASTIC LEAVES 1 (HYL1), and the zinc-finger protein Serrate (SE)[3−5]. Next, the nascent miRNA/miRNA* duplex undergoes 2’-O-methylation at the 3’ ends by HUA ENHANCER1 (HEN1)[6]. In Arabidopsis, mutants with complete miRNA biogenesis defects are embryo-lethal. Embryos lacking DCL1 arrest early in development and exhibit aberrant patterning in most regions of the embryo[7,8]. Similarly, embryos of serrate and hyl1 also show aberrant phenotypes at the beginning of the zygote development[9]. These abnormal phenotypes in an embryo could be partially attributed to the ovule development defects and reduced female fertility caused by mutations of miRNA biogenesis factors[10,11]. However, whether and to what extent the miRNA biogenesis factors in male gametophytes contribute to male fertility and early seed development are still not well understood.
In flowering plants, the male gametophyte microspores undergo two rounds of mitosis. The first asymmetric mitotic division (PMI) produces two daughter cells with different cell fates: a larger vegetative cell (VC) and a smaller generative cell (GC). The VC exits its cell cycle while the GC undergoes another mitotic division is known as pollen mitosis II (PMII) to produce two sperm cells (SCs). This process results in a mature pollen with the two sperm cells embedded in the cytoplasm of the vegetative cell[12]. Increasing evidence has revealed the extreme heterogeneity between two cell types in mature pollen, characterized by distinct epigenetic controls[13−17]. Interestingly, substantial communication persists between the VC and the GC/SCs, particularly involving the movement of small RNA reported during pollen development[18−22]. miRNAs also play a crucial role in these processes by regulating gene and transposable element (TE) transcripts. It has been reported that pollen miRNAs serve dual functions: one is to target genes related to pollen development and germination, while the other is to mediate the biogenesis of TE-derived secondary epigenetically activated siRNAs (easiRNAs), thereby initiating sperm silencing of TEs[23−25]. One typical example of the latter is the accumulation of miR845-directed 21, 22-nt easiRNAs in a dose-dependent manner, which mediates hybridization barriers between diploid seed parents and tetraploid pollen parents (triploid block)[26].
The complexity of pollen development and the communication between two types of cells highlight the distinct biological functions of miRNA biogenesis in the VC and the GC/SC. In this study, we explored the spatiotemporal pattern of miRNA biogenesis across various stages of pollen development, focusing on the dynamics of MIR gene transcription and the distribution of miRNA biogenesis factors. Using an artificial miRNA strategy driven by cell type-specific promoters, DCL1 was selectively knocked down in the VC and GC/SC. Our findings underscored the critical roles of miRNA biogenesis in these two cell types of pollen, emphasizing their contributions to fertility control and seed development.
-
Columbia-0 (Col-0) ecotype was used in this study as wild-type plants. The Arabidopsis mutant lines used in this study, including dcl1-7 (CS3089) and the myb33 myb65 double mutant created by introducing a Cas9 deletion of MYB65 into the myb33 background (CS851168), are all in the Col-0 background. The reporter lines of proMIR845a::GFP, proMIR845b::GFP, proMIR164a::GFP, and proMIR2939::GFP were constructed by our lab, the proMIR159a::GFP and proMIR159b::GFP lines were described in a previous study[27]. Transgenic plants of proVCK1::amiR_DCL1 and proHTR10::amiR_DCL1 were generated in this study. All plants were grown in the soil with a humidity of 65% under a 16 h light/8 h dark photoperiod at 22 °C.
Plasmid construction and plant transformation
-
To generate the constructs of MIRNA reporter lines of proMIR845a::GFP, proMIR845b::GFP, proMIR164a::GFP, and proMIR2939::GFP, the promoter region of MIRNA genes was amplified from Col-0 genomic DNA, cloned into pENTR-D/TOPO, and then transferred into proGW::NLS-2 × GFP by LR reaction. For proVCK1::amiR_DCL1 and proHTR10::amiR_DCL1, the artificial miRNA were designed by the WMD3 website (
http://wmd3.weigelworld.org ) , and the amiR_DCL1 fragments were amplified from the pRS300-miR319a backbone and cloned to pB7WG2-HTR10 or pB7WG2-VCK1. For myb65-cas9 mutant, the web tool CHOPCHOP (http://chopchop.cbu.uib.no ) was used to design the sgRNAs. The primers of MYB65-DT1-BsF, MYB65-DT1-F0, MYB65-DT2-R0, MYB65-DT2-BsR were used to amplify the fragment using plasmid pCBC-DT1T2 as a template, then the amplified fragment was digested with BsaI and inserted into pHEE401E. All primers are listed in Supplementary Table S1.Phenotypic analysis
-
Flowers at stage 12 were emasculated and pistils were left to grow for ~12 h for maturation. Then pistils were hand-pollinated with pollen grains of Col-0 or the dcl1-7 mutant. To measure the length and seed number of F1 siliques, and pistils at 7 days after pollination (DAP) were dissected and the seed numbers were counted under a Leica dissecting microscope. To calculate the proportion of viable seeds in different T1 lines of the transgenic plants, and self-pollinated pistils at 8 DAP were dissected under a microscope. To observe the pollen tubes growth in vitro, pollen grains were spread on the solid pollen germination medium (1 mM CaCl2, 1 mM Ca(NO3)2, 1 mM MgSO4, 1.62 mM H3BO3, 18% (w/v) sucrose, 1% (w/v) low melting agarose, adjusted to pH 7.0 with 0.1 M KOH) and incubated in a humid chamber at 28 °C for 6 h[28]. To examine embryo development with differential interference contrast (DIC) microscopy, the seeds at 7 DAP were mounted in clearing solution (chloral hydrate : water : glycerol, w/v/v, 8:3:1) for DIC imaging with UPlanFLN × 20 objective. Images were further processed using Adobe Photoshop and Image J.
Microscopy
-
To visualize the promoter-reporter of MIRNA genes, fluorescence microscopy analysis was carried out with an Olympus BX53 microscope (image acquisition software: QCapture Pro7; objectives: UPlanFLN 40×). DAPI staining of pollen was examined under a UV channel using UPlanFLN 40 × objectives. To visualize the localization of miRNA biogenesis factors, pollen grains were observed by the Leica Stellaris 5 WLL confocal microscope with the lightning mode. Images were processed using LAS X and Image J.
Statistical analysis
-
All raw data were imported into GraphPad Prism version 9.5 (GraphPad Software) and then analyzed by Student’s t-test, one-way ANOVA, or Chi-square test. A value of p < 0.05 was considered as statistically significant. ****, ***, **, and * indicate p < 0.0001, 0.001, 0.01, and 0.05, respectively. ns, no significant difference.
-
It has been reported that several dcl1 mutants, such as dcl1-3, dcl1-4, dcl1-5, dcl1-10, dcl1-7, and dcl1-15, display abnormalities in embryo development[9]. Moreover, maternal defects of several dcl1 alleles significantly contribute to the defects of embryo development[10,11]. However, it has been shown that paternal DCL1 from wild-type pollen can partially rescue the embryo-lethal phenotype when dcl1-5/+ mutants are used as females[29]. This finding indicates that paternal DCL1-mediated miRNA biogenesis potentially contributes to embryo development. In this study, a weak allele, dcl1-7 (CS3089) was used, which allowed for the production of viable homozygous mutant plants, to perform the hand-pollination assay. The dcl1-7 homozygous plants displayed defects in anther development (Supplementary Fig. S1a) and a large proportion of pollen grain with low viability (Supplementary Fig. S1b). Notably, at least 50 pollen grains per anther retained normal viability. Furthermore, DAPI staining revealed that these viable pollen grains in the dcl1-7 mutant contained a vegetative nucleus and two sperm nuclei (Supplementary Fig. S1c & d, also shown below), similar to wild-type pollen grains. This suggests that the non-viable pollen grains are likely a result of impaired anther (sporophyte) development, rather than defects in male gametophyte development itself. Given that some pollen grains remained viable in the dcl1-7 mutant, we aimed to investigate whether paternal miRNA biogenesis plays a role in subsequent processes such as double fertilization and early seed development.
To investigate this question, we assessed seed development by pollinating wild-type pistils with excess pollen from both Col-0 wild-type pollen and the dcl1-7 mutant pollen. Compared with F1 siliques from ♀Col-0 × ♂Col-0 crosses, F1 siliques from ♀Col-0 × ♂dcl1-7 crosses showed significantly shortened siliques (Fig. 1b) and severe defects in seed development, resulting in only 20%−30% viable seeds (Fig. 1c). These observations indicate that paternal miRNA biogenesis machinery in mature pollen contributes to subsequent seed development.
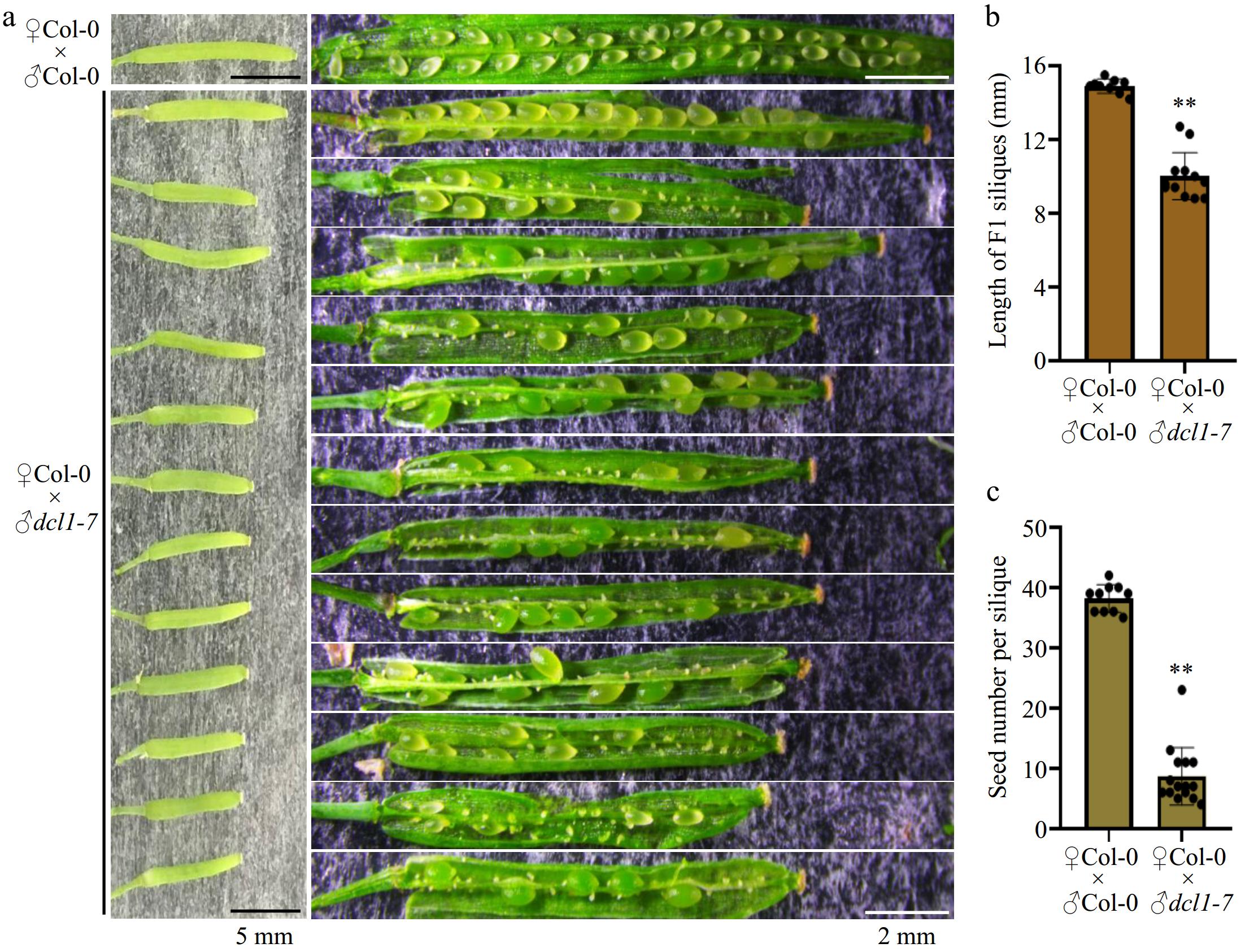
Figure 1.
Hand-pollination by the dcl1-7 mutant pollen caused defects in seed production. (a) Representative F1 siliques from ♀Col-0 × ♂Col-0 and ♀Col-0 × ♂dcl1-7 respectively. The dcl1-7 mutant pollen as the male causes aborted seeds. Black bars indicate 5 mm length, and the white bars indicate 2 mm length. (b), (c) Statistical analysis showing the (b) length, and (c) the total number of the F1 siliques. Each point indicates one silique, p-value were calculated using an unpaired, two-sided Student t-test (** p < 0.01). Scale bars are shown.
To conclusively demonstrate these results, we followed our previous study, where sperm-delivered miR159 was shown to promote seed development by removing maternal roadblock targets[30]. Given the developmental defects observed from early vegetative growth stages in the mir159abc triple mutant, we investigated the contribution of paternal-generated miR159 to the phenotypes of shortened siliques and aborted seeds. Consistent with the male germ cell-specific expression of HTR10[31], the over-expression of MIR159a driven by the HTR10 promoter did not complement the vegetative growth phenotypes (Supplementary Fig. S2a), but partially rescued the defects on silique length and seed setting rate of the mir159abc triple mutant (Supplementary Fig. S2b−d). The defects in endosperm nuclear divisions fertilized by miR159abc pollen were attributed to the retention of miR159 targets MYB33 and MYB65 in the central cell after fertilization[30]. Moreover, the decreased proportion of viable seeds in F1 siliques obtained by mir159abc pollen as male could be restored when using the myb33 myb65 double mutant as female instead of Col-0 (Supplementary Fig. S2e−g), although the myb33 myb65 double mutant exhibited partial shortened silique phenotype in self-pollination (Supplementary Fig. S2f). These results further confirmed our previous observation that sperm-delivered miR159 promotes seed development by repressing maternal MYB33 and MYB65. Collectively, it was conclusively demonstrated that paternal miRNA biogenesis is essential for fertilization and viable seed production.
MIR genes are preferentially transcribed at early stages of pollen development
-
A recent study has analyzed the miRNA profiles at the microspore, bicellular pollen, and mature pollen stages, thereby providing an atlas of the miRNome during pollen development[25]. The overall levels of total miRNAs are similar among microspore, bicellular pollen, and tricellular pollen stages, but slightly increased in mature pollen (Supplementary Fig. S3a). This suggests that MIR transcription and/or miRNA biogenesis may not exhibit significant dynamics during pollen development. To investigate whether this is the case, the timing and location of MIR gene transcription during pollen mitoses was first examined, focusing on several well-known abundant miRNAs in mature pollen. Several transgenic lines with GFP reporters driven by endogenous promoters of pollen-enriched miRNAs were constructed, including MIR159a, MIR159b, MIR164a, MIR845a, MIR845b, and MIR2939. The present findings show that all these MIR genes initiated their transcription primarily early in the microspore stage before PMI (Fig. 2a). Following PMI, transcription of these tested MIR genes continued in both VC GC, with a slightly stronger signal observed in the VC-based on GFP intensity (Fig. 2b). However, the transcriptional signal of five out of the six tested MIR genes was completely absent in both VC and GC at the mature pollen stage (Fig. 2c). The exception was proMIR2939::GFP, which exhibited weak but robust expression signals in sperm cells (Fig. 2c). Considering that all four miRNAs are relatively steadily produced across pollen development (Supplementary Fig. S3b)[25], the phenomena of MIR transcription at early stages of pollen development indicate that miRNAs detected in mature pollen are obtained by either local processing of early transcribed pri-miRNA or inherited from bicellular pollen via PMII.
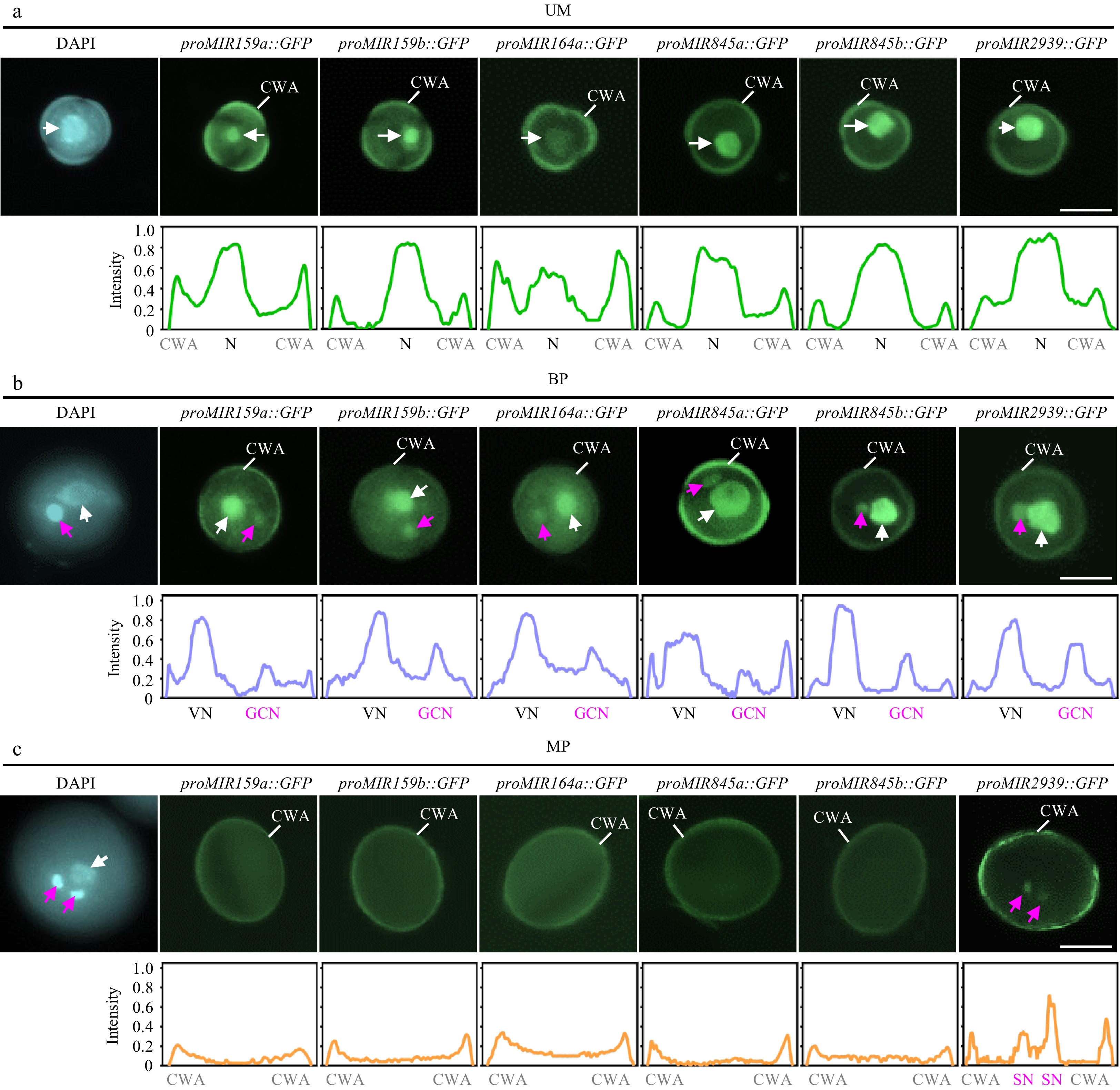
Figure 2.
Spatiotemporal patterns of MIR transcription during pollen development. The expression of proMIR159a::GFP, proMIR159b::GFP, proMIR164a::GFP, proMIR845a::GFP, proMIR845b::GFP, and proMIR2939::GFP was observed during pollen development by fluorescence microscopy. Representative pollen grains show DAPI fluorescence (left, blue) and GFP fluorescence (right, green) at unicellular microspore [(a), UM], bicellular pollen [(b), BP], and mature pollen [(c), MP] stages. White arrowhead indicates N or VN; magenta arrowhead indicates GCN or SN. Relative intensity curve of the GFP signal were shown below the images. VN, vegetative nucleus; GCN, generative cell nucleus, SN, sperm cell nuclei; CWA, cell wall autofluorescence. Scale bar = 10 μm.
miRNA biogenesis factors are ubiquitously expressed during pollen mitosis
-
Since the transcription of MIR genes mainly occurs before the second pollen mitosis, does miRNA biogenesis also exhibit similar properties? RNA-seq data from the vegetative cells and sperm cells of mature pollen were first analyzed[32], focusing on the expression of several main factors involved in miRNA biogenesis. The results show that while DCL1, SE, and HEN1 are detectable in both sperm cells and the vegetative cell, the overall mRNA levels of these genes are low in mature pollen (Supplementary Fig. S3c). Notably, mRNA of HYL1 is barely detected in mature pollen (Supplementary Fig. S3c). These analyses indicate that the local activity of the intact miRNA biogenesis machinery in mature pollen is low.
To further elucidate the spatiotemporal pattern of miRNA biogenesis during pollen mitosis, we investigated the subcellular localization of DCL1, HYL1, and HEN1 throughout pollen development using corresponding transgenic plants expressing DCL1-YFP, HYL1-YFP, and HEN1-YFP, each driven by their native promoters[33]. The results show that all three proteins are highly detected in the microspore (Fig. 3a−d, top panels), indicating the potential for local processing of pri-miRNA. After the first pollen mitosis, there were no significant differences in DCL1-YFP, HYL1-YFP, and HEN1-YFP signal intensity between the vegetative cell and the generative cell at the bicellular pollen stage (Fig. 3a-d, the middle panels). However, the intensities of DCL1-YFP and HYL1-YFP were decreased in sperm cells at the mature pollen stage, while the intensity of HEN1-YFP in sperm cells remained at the same level as in the vegetative cell (Fig. 3a−d, the middle panels). These results indicate that miRNA biogenesis factors persist in the vegetative cell throughout pollen development. Instead, miRNA biogenesis activity may be compromised in sperm cells due to a much lower accumulation of DCL1, suggesting that the miRNAs enriched in sperm cells may be inherited from bicellular pollen or transferred from the vegetative cell at the mature pollen stage. The high signal of HEN1-YFP suggests that sperm-enriched small RNAs may require HEN1-mediated 3’ end methylation. Notably, while the miRNA biogenesis machinery has been found to localize in dicing bodies, no nuclear punctate structures were observed for all three proteins in pollen. This indicates that the subcellular territory of miRNA biogenesis in pollen may differ from that in somatic cells.
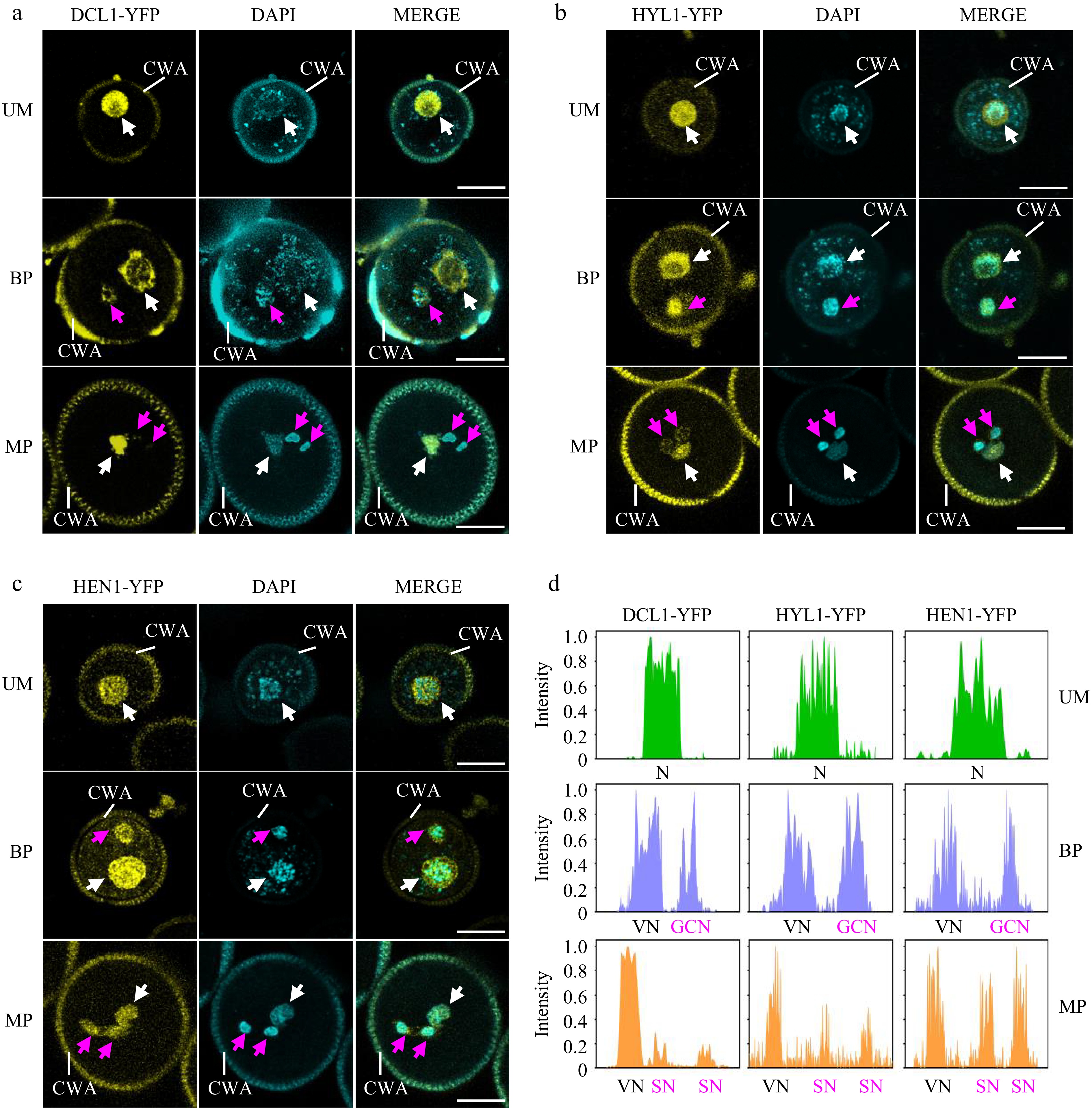
Figure 3.
Spatiotemporal patterns of miRNA biogenesis factors during pollen development. (a)−(c) The localization of (a) DCL1-YFP, (b) HYL1-YFP, and (c) HEN1-YFP was observed during pollen development by confocal microscopy. Representative pollen grains show YFP fluorescence (right, yellow) and DAPI fluorescence (right, cyan) at unicellular microspore (UM), bicellular pollen (BP), and mature pollen (MP) stages. White arrowhead indicates N or VN; magenta arrowhead indicates GCN or SN. VN, vegetative nucleus; GCN, generative cell nucleus, SN, sperm cell nuclei; CWA, cell wall autofluorescence. Scale bar = 10 μm. (d) Relative intensity curve of YFP signal in (a) DCL1-YFP, (b) HYL1-YFP, and (c) HEN1-YFP.
Conditional knockdown of DCL1 in VC and GC/SC caused compromised but distinctive defects in seed development
-
As the transcription of MIR genes is mostly completed before the second pollen mitosis (Fig. 2), what is the biological significance of the persistence of the miRNA biogenesis machinery in mature pollen? To investigate the role of miRNA biogenesis factors detected in the vegetative cell and sperm cells of mature pollen, two types of transgenic lines were generated with cell-type-specific knockdown of DCL1 in the VC and GC/SC, respectively. Expression of the artificial miRNA targeting DCL1 was achieved by using the late VC-specific promoter VCK1[34] and the GC/SC-specific promoter HTR10[31], respectively. These constructs were transformed into the proDCL1::DCL1-YFP reporter line to primarily evaluate the efficiency of knockdown by the artificial miRNA (Supplementary Fig. S4a). As shown in Supplementary Fig. S4a, the introduction of the VC-expressed artificial miRNA targeting DCL1 significantly attenuated the signal intensity of DCL1-YFP in the vegetative cell. Similarly, the introduction of the GC/SC-expressed artificial miRNA targeting DCL1 specifically attenuated the signal intensity of DCL1-YFP in the sperm cells, while having no effect on the signal in the vegetative cell. When proVCK1::amiR_DCL1 was introduced, the faint signal of DCL1 in the vegetative nucleus could be still observed, indicating a possible leaky expression. Whether the reduced expression of DCL1 in the vegetative cell and sperm cells affects double fertilization and subsequent seed development were then examined. In eight independent transgenic T1 lines expressing proVCK1::amiR_DCL1 in the vegetative cell, we found that these plants exhibited noticeably shortened siliques, reduced seed numbers, and approximately half of the seeds were unfertilized (Fig. 4a, c), which resembled the phenotypes observed in the F1 silique of ♀Col-0 × ♂dcl1-7 (Fig. 1a). Interestingly, in ten individual transgenic T1 lines expressing proHTR10::amiR_DCL1 in the generative cell and sperm cells, it was observed that approximately 30% of seeds exhibited a typical embryo lethal phenotype (Fig. 4b). Additionally, these plants produced an average of about 30 seeds per silique (Fig. 4d), compared to the Col-0 plants, which produce approximately 50 seeds per silique on average. These results indicating that DCL1 expressed in sperm is involved in early embryogenesis after double fertilization.
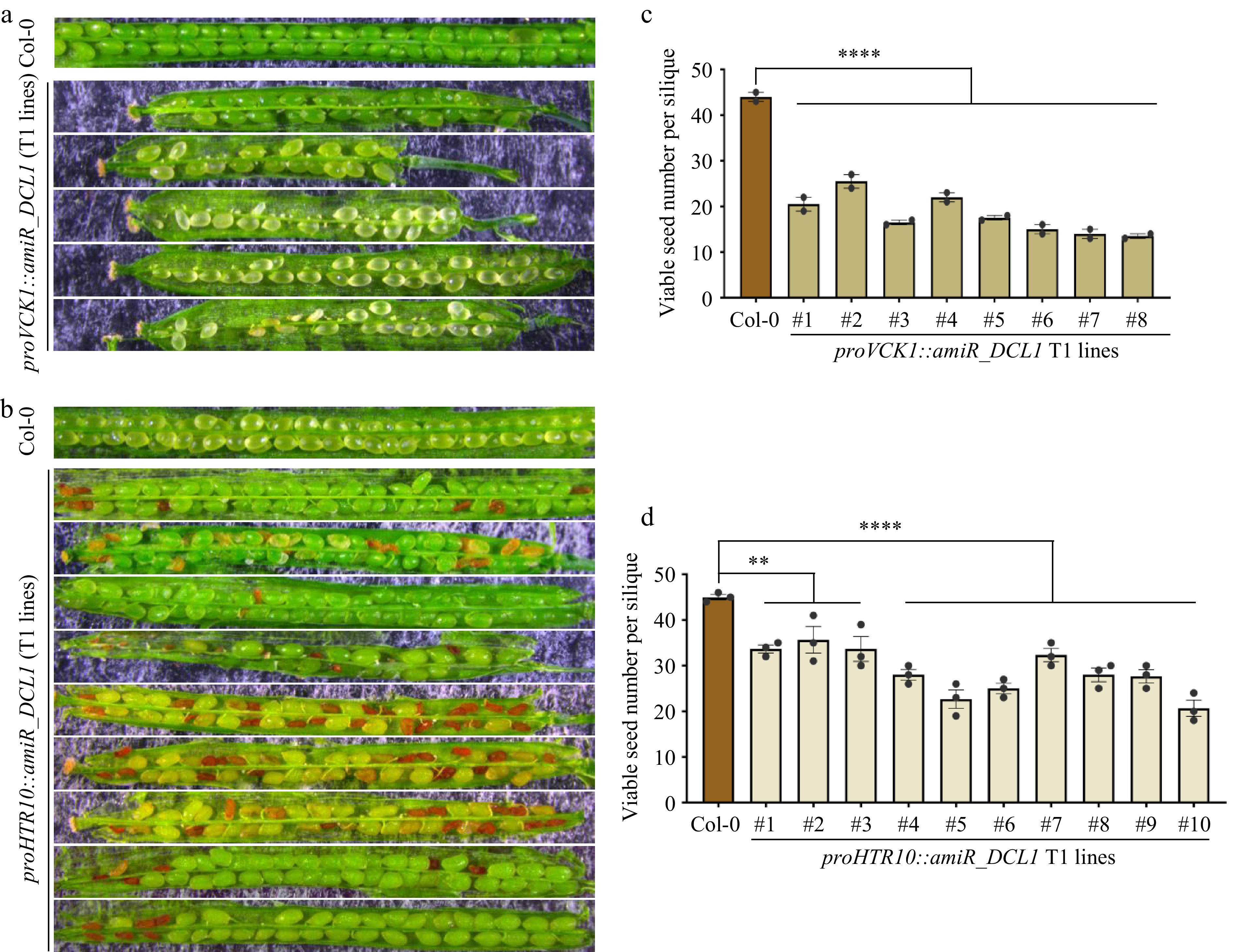
Figure 4.
Both VC and GC/SC-knockdown of DCL1 caused compromised but distinctive defects in seed development. (a) Seed development defects of proVCK1::amiR_DCL1 transgenic T1 lines, representative siliques are shown. (b) Seed development defects of proHTR10::amiR_DCL1 transgenic T1 lines, representative siliques are shown. (c), (d) Statistical analyses of viable seed number per silique in (c) proVCK1::amiR_DCL1, and (d) proHTR10::amiR_DCL1 transgenic T1 lines. Each column indicates one individual T1 line. p-values were calculated by one-way ANOVA Dunnett’s multiple comparison, ** p < 0.01, **** p < 0.0001.
Given the nature of phenotypes in seed development of plants with conditional knockdown of DCL1 in mature pollen, we hypothesized that the seed development defects in proVCK1::amiR_DCL1 plants should be attributed to the fertilization failure rather than post-fertilization impairment. To test this, the in vitro pollen germination rate of Col-0 and proVCK1::amiR_DCL1 heterozygous plants were preliminarily assessed. Compared to Col-0, five transgenic lines of proVCK1::amiR_DCL1 showed a significantly reduced pollen germination rate (Supplementary Fig. S4b & c), indicating that VC-expressed DCL1 plays an important role in pollen germination. In contrast, the transgenic lines expressing proHTR10::amiR_DCL1 exhibit approximately 30% aborted seeds (Fig. 4b & d). To further investigate the arrested stage of the abnormal seeds in the proHTR10::amiR_DCL1 transgenic lines, we observed the embryo development of normal and abnormal seeds in a same proHTR10::amiR_DCL1 silique at 7 DAP, when the abnormal seeds had not been completely dried out. DIC microscopy analyses showed that the embryos of the abnormal seeds were arrested at the globular stage, while mature embryos had been developed in the normal seeds of the same silique (Supplementary Fig. S4d). The arrest of embryos at the globular stage is consistent with the embryo abnormalities observed in several dcl1 mutant alleles[7−9,29], demonstrating that paternal DCL1 contributes to early embryo patterning during seed development. Together, it is concluded that mature pollen-expressed DCL1 is necessary for pollen germination and early embryo development.
-
The embryo lethal phenotype of dcl1 mutant alleles highlights the remarkable role of miRNA biogenesis during seed development[8,9,29]. Due to the size discrepancy between sperm cells and the egg cell, it is generally believed that substances in egg cells play a major role in early development. However, it is increasingly clear that the male gamete carries additional coding or non-coding RNAs necessary for early development in different species[35,36]. Our previous work shows that paternal miR159 is required to promote endosperm nuclear divisions during early seed development[30]. In this study, we further demonstrate that mature pollen-expressed miRNA biogenesis factors contribute to both the fertilization process and early embryo development.
The spatiotemporal dynamics of MIR transcription and miRNA biogenesis factors through pollen mitosis indicate that the biogenesis of most pollen-enriched miRNAs is completed at early developmental stages before PMII. Furthermore, the miRNA biogenesis patterns between the vegetative cell and male germ cells exhibit both similarities and differences, supported by these observations: (i) pollen-expressed MIR genes preferentially complete their transcription before the second pollen mitosis in both the vegetative cell and the generative cell; (ii) miRNA biogenesis factors are localized in both the vegetative cell and generative cell at early developmental stages; (iii) the abundance of DCL1 shows a striking decrease in sperm cells compared to the vegetative cell. The differences in the dicing complex between the vegetative cell and sperm cells might be attributed to the de-condensed chromatin in the vegetative cell and the gradually increasing chromatin condensation in sperm cells[37]. Notably, previous study on miRNA profiling in vegetative and sperm cells have shown that specific miRNAs are distinctly enriched between these two cell types[38], indicating that miRNA distribution within mature pollen is tightly regulated. Based on the vegetative cell-preferred localization of DCL1 and the transcription of MIR genes before sperm cell formation, the specificity of miRNA distribution in the two cell types within mature pollen may not be determined by the localization of miRNA biogenesis machinery. Instead, likely specific miRNAs are actively transferred from the vegetative cell to the sperm cells[18−21]. One typical example is miR159, where the transcription of three MIR159 genes occur before the second pollen mitosis, but mature miR159 is significantly enriched in sperm cells[38].
Unlike specific miRNAs that function in pollen by inhibiting target genes expressed either in pollen itself or on the maternal side, the fact that reduced expression of DCL1 in both the vegetative cell and sperm cells causes distinct defects in seed development indicates that mature pollen-expressed DCL1 is important for specific miRNA biogenesis in pollen and the pollen tube, or is delivered into female gametes via fertilization to promote early miRNA biogenesis. The effects of vegetative cell-expressed DCL1 on pollen germination and sperm cell-expressed DCL1 on embryo development align with the default functions of the vegetative cells and sperm cells, respectively. The present data showed that a significantly reduced pollen germination rate in vegetative-cell dcl1 knockdown plants, indicating that the seed development defects in proVCK1::amiR_DCL1 plants are likely due to fertilization failure rather than post-fertilization impairment. We suggest that DCL1-mediated miRNA biogenesis in the vegetative cell may also play a role in pollen tube targeting or other processes required for successful double fertilization. Meanwhile, the embryo development defects in proHTR10::amiR_DCL1 transgenic lines resemble the embryo abnormalities in dcl1 mutants[9,29], indicating that not only maternal but also paternal DCL1 contributes to early embryo patterning.
This work was supported by the National Natural Science Foundation of China (32025005, 31830045, M-0398 to BZ).
-
The authors declare that they have no conflict of interest. Binglian Zheng is the Editorial Board member of Seed Biology who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and the research groups.
- Supplementary Table S1 Primer sequence.
- Supplemental Fig. S1 The dcl1-7 homozygous plant showed disrupted anther development but normal pollen mitoses.
- Supplemental Fig. S2 Male germ cell-overexpressed MIR159a partially rescued fertility defects of the mir159abc mutant.
- Supplemental Fig. S3 miRNA levels and expression of main miRNA biogenesis factors are adapted from publicly available data during pollen development.
- Supplemental Fig. S4 Pollen germination and embryo development in conditionally knockdown plants of dcl1 in pollen.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Hainan Yazhou Bay Seed Laboratory. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yang H, Zhao Y, Lin Z, Jiang T, Hu Q, et al. 2024. Paternal miRNA biogenesis contributes to seed development in Arabidopsis. Seed Biology 3: e017 doi: 10.48130/seedbio-0024-0017 

Paternal miRNA biogenesis contributes to seed development in Arabidopsis
- Received: 25 July 2024
- Revised: 25 September 2024
- Accepted: 21 October 2024
- Published online: 30 October 2024
Abstract: miRNAs are key regulators of gene expression and play important roles in various developmental processes. The development of the plant male gametophyte begins with a microspore, which undergoes two rounds of pollen mitosis to produce mature pollen, consisting of a vegetative nucleus and two sperm cells. Although many miRNAs are known to accumulate in mature pollen, it remains unclear how miRNA biogenesis is regulated during pollen mitosis and whether miRNA biogenesis in mature pollen is necessary for seed development. Here, we focus on DCL1, the major enzyme for miRNA biogenesis in Arabidopsis. Hand-pollination using dcl1-7 mutant pollen results in severe seed development defects, characterized by shortened siliques and approximately 80% aborted seeds. While miRNA genes are primarily transcribed at early stages of pollen development, the core factors of the miRNA biogenesis machinery are expressed throughout pollen development, with preferential expression in the vegetative nucleus. Using an artificial miRNA strategy to conditionally knock down DCL1 in the vegetative cell and sperm cells, respectively, we demonstrate that miRNA biogenesis in both cell types contributes to fertility control, but results in distinct defects in seed development. Collectively, these results show that miRNA biogenesis in mature pollen plays a significant role in regulating fertility and seed development.
-
Key words:
- Pollen mitosis /
- miRNA biogenesis /
- DCL1 /
- Seed development.