






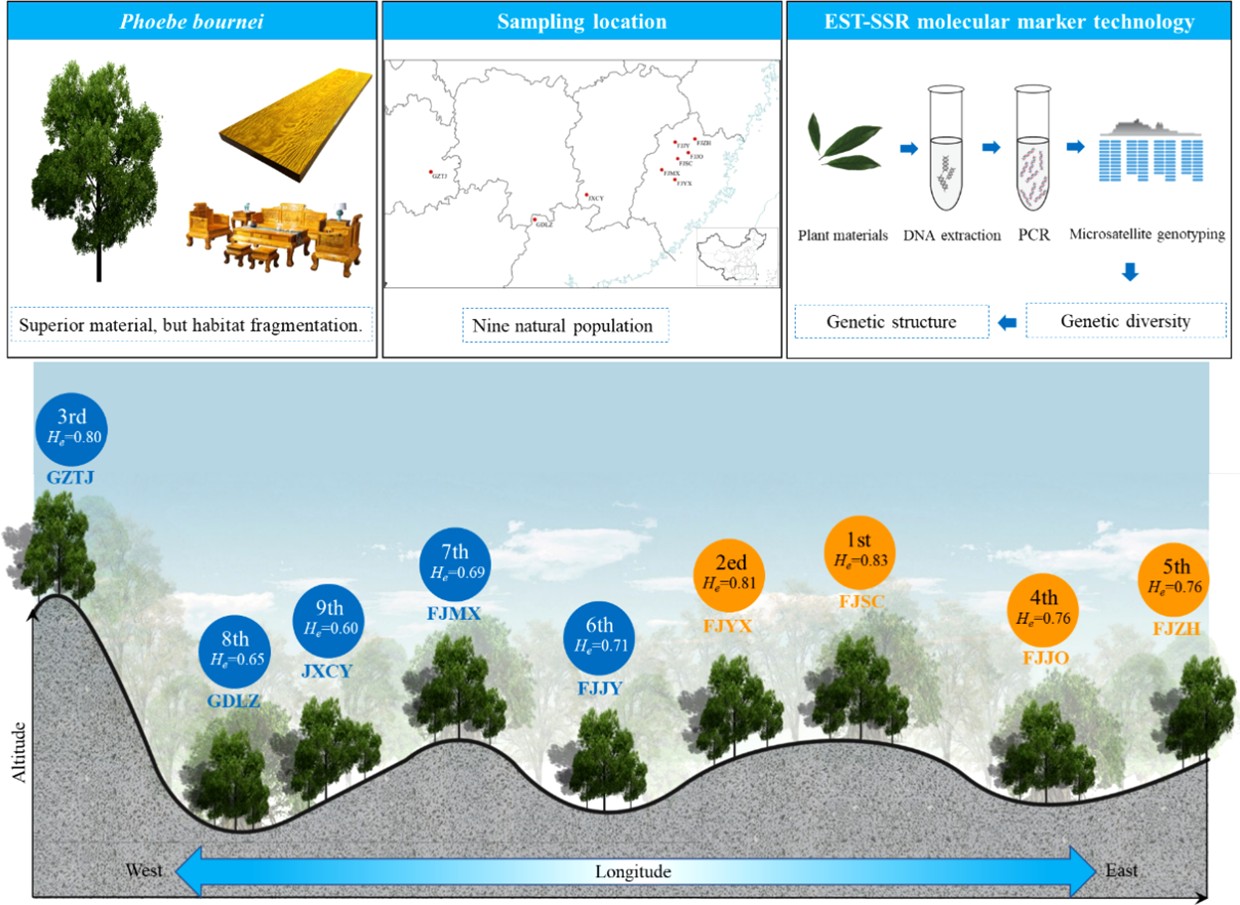


-
The genetic diversity of a species is determined by the sum of the genetic differences between individuals. The emergence of new alleles in each generation is caused by DNA damage due to replication errors or mutagenesis. The presence and absence of genetic variations reflect the balance of genetic diversity, which is an essential basis for the sustainable survival and development of species in complex environments. Environmental adaptation during long-term species evolution shapes genetic diversity in populations[1]. In an ideal population, random mating between individuals, gene frequency, and genotype frequency remain consistent across generations[2]. However, real populations typically deviate from these assumptions. Factors such as population size[3], age structure[4], gender ratio[5], mating system, and seed dispersal mode[6] all affect reproductive and natural regeneration abilities, as well as the survival potential of endangered species, thereby influencing the maintenance of genetic diversity[7,8]. In addition, genetic diversity is inversely proportional to population size, where small populations and great spatial isolation caused by habitat fragmentation result in severe genetic drift, inbreeding, and the accumulation of harmful mutations. Ultimately, these factors lead to the low genetic diversity of populations and even extinction[9].
Thus, evaluating genetic diversity is crucial for protecting and utilizing germplasm resources and genetic breeding. Such a procedure significantly reveals the genetic structure, evolutionary history, potential, and causes of species endangerment. Additionally, this evaluation provides predictive guidance for selecting parents, determining the degree of genetic variation in offspring, and assessing the level of heterosis.
Simple sequence repeats (SSRs) are widely distributed in the genomes of eukaryotes and are a more accurate way to study species diversity than using phenotype variations. SSR molecular marker technology has several characteristics, such as co-dominance, high resolution, and good repeatability making it one of the most widely used molecular marker technologies in plant genetic diversity analysis[10]. However, the silver-staining technology used during the experimental process may impact the accuracy of the identification of different allele genes. Capillary electrophoresis technology overcomes the disadvantages of traditional silver staining methods, allowing for automated processing and data collection of large-scale samples SSR analysis, which improves the reliability of the markers. Neophytou et al. used SSR molecular marker technology to discover a significant decrease in the genetic diversity of adult Pseudotsuga menziesii trees compared to natural regeneration seedlings[11]. Wu et al. found low genetic diversity within Glyptostrobus pensilis populations based on EST-SSRs[1]. Genetic differentiation among populations was substantial, suggesting that long-term geographical isolation may be the primary reason for this regional genetic pattern.
Phoebe bournei is a broad-leaved, evergreen tall tree belonging to the family Lauraceae in the genus Phoebe. It is related to the commercial species, Phoebe zhennan, and is a rare and endangered tree species at the national second-level protection. The wood of this species is tough, dense, and has beautiful patterns, making it a superior material for architecture, high-end furniture, and craft carving[12]. This species is also a rare and unique ornamental tree in China. P. bournei is named after the abbreviation of Fujian province, where its natural population is mainly distributed. However, it is also found in other provinces (autonomous regions), such as Zhejiang, Jiangxi, Hunan, Guangdong, Guangxi, and Guizhou. P. bournei is widely distributed and has an extensive range and considerable geographical distance between populations. Due to human activities, P. bournei habitat fragmentation has been severe for a long time, and the natural germplasm resources are scarce.
There are still fewer studies on the genetic diversity of natural populations of P. bournei based on SSR molecular markers on a large scale. The natural populations and sample sizes involved in previous studies are small, the distribution range is narrow, the geographical distance between populations is close, and there is a lack of in-depth systematic studies, which are not sufficient to comprehensively reveal the genetic diversity and genetic structure of natural populations of P. bournei[13]. Therefore, this study selected six natural populations of P. bournei in the Fujian province and three in other provinces as materials. The genetic diversity and structure of natural populations of P. bournei were systematically revealed based on EST-SSR molecular marker technology, which provides a theoretical basis for the protection of germplasm resources and the exploration and utilization of excellent germplasms of P. bournei.
-
Nine natural populations of P. bournei were selected from its central distribution areas in China from November to December 2021, including six in Fujian Province and one each in Chongyi in Jiangxi Province, Taizhou in Guizhou Province, and Lianzhou in Guangdong Province (Fig. 1). At least 50 evenly distributed samples were collected form each population. Populations with less than 50 samples were collected entirely, resulting in the collection of 460 single plants. Details for each population are shown in Table 1. Five to ten fresh tender leaves were collected from each plant and placed in sealed bags, then stored in a refrigerator at −20 °C and transported to the laboratory for DNA extraction. The longitude, latitude, and elevation of the location of each plant were recorded using a GPS device, and the diameter at breast height was measured.
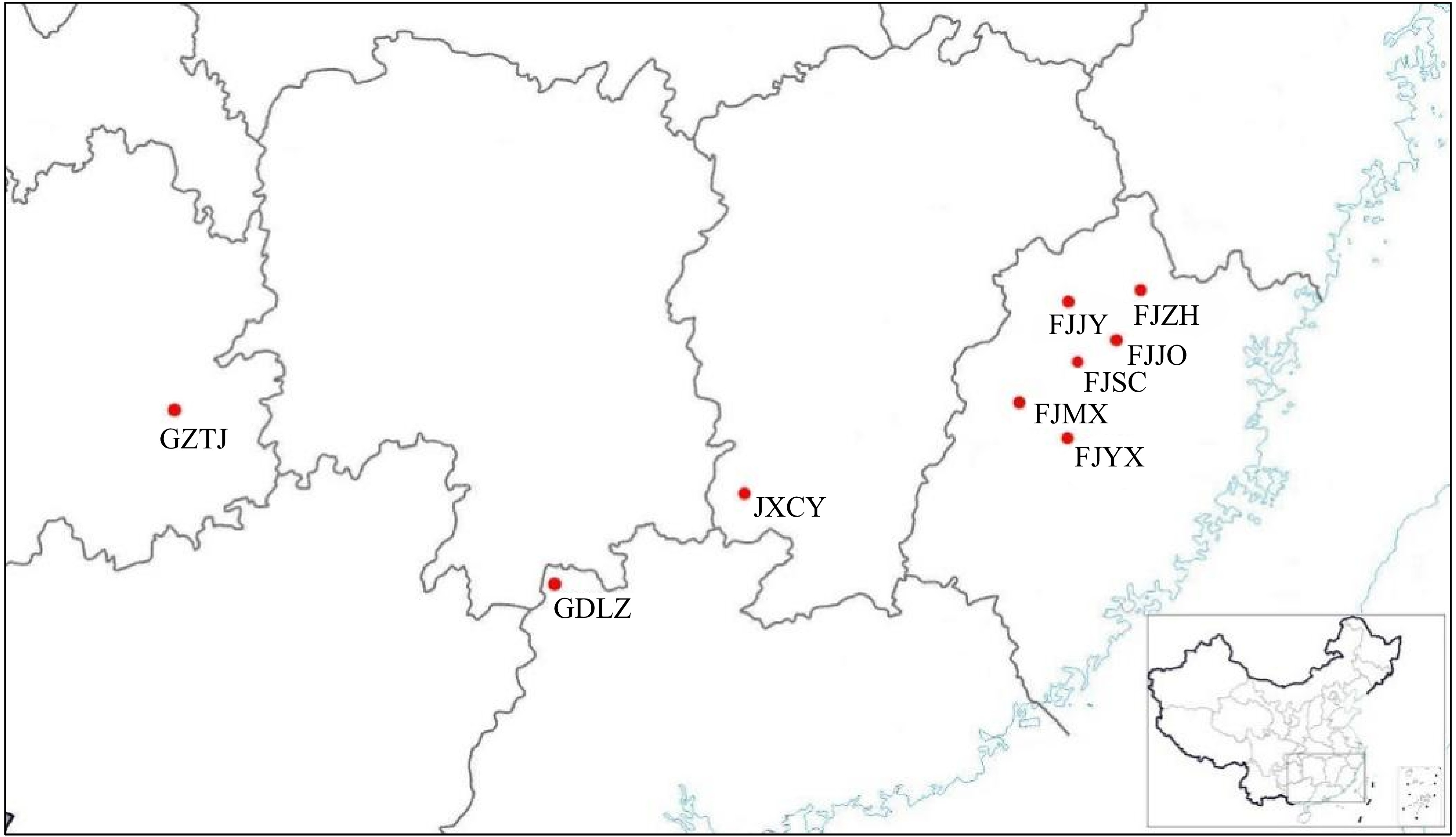
Figure 1.
Sampling locations of the natural populations of P. bournei.
Table 1. Sample numbers and geographic information for experimental analysis of nine natural populations of P. bournei.
Serial number Population code Location Sample numbers Longitude (E) Latitude (N) Altitude (m) 1 FJJO Jian'ou, Fujian 28 118.37 27.08 148.16 2 FJJY Jianyang, Fujian 50 117.84 27.39 197.82 3 FJMX Mingxi, Fujian 56 117.27 26.41 442.37 4 FJSC Shunchang, Fujian 69 117.94 26.87 434.72 5 FJYX Youxi, Fujian 55 118.28 26.27 308.68 6 FJZH Zhenghe, Fujian 50 118.62 27.44 299.60 7 GDLZ Lianzhou, Guangdong 50 112.20 25.05 195.85 8 GZTJ Taijiang, Guizhou 50 108.31 26.58 802.62 9 JXCY Chongyi, Jiangxi 52 115.17 26.36 219.98 DNA extraction and microsatellite genotyping
-
A DNA extraction kit was used to extract the genomic DNA of P. bournei. After that, 1% agarose gel electrophoresis was used to verify the integrity of the DNA. Next, a NanoDrop spectrophotometer was employed to assess the concentration and purity of the DNA, aiding in subsequent PCR amplification.
Based on the literatures[14,15], 16 pairs of EST-SSR primers with clear bands and substantial polymorphisms were selected for PCR amplification (Table 2). The reaction system was 25 μl : 2 μl DNA template, 1 μl forward primer, 1 μl reverse primer, 12.5 μl PCR Master Mix, and 8.5 μl ultrapure water. The DNA PCR amplification was performed using an ABI Veriti amplification instrument, with the following reaction program: predenaturation at 94 °C for 5 min, denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s, extension at 72 °C for 30 s, for 35 cycles. The final extension was performed at 72 °C for 10 min. PCR products were detected using a fully automated nucleic acid protein analyzer (Qsep-100), and Q-Analyser-for-100 software was used to identify gene fragments.
Table 2. Characteristics of 16 SSR loci.
Locus Primer sequence (5'-3') Length (bp) Repeat motif Annealing temperature (°C) Ref. L1 F: TCGATTTGCAGAAGATAAGCC 449 (ATT)14 63 [15] R: GGGGTAGAAAAGTGAAAGAGTTG L2 F: AGAGGGCCTGTGCGTACGTTT 352 (TCT)12 63 [15] R: ACATTTGAGTCGGTTCCGGTTCC L3 F: GCTAGAGCTCAAAGGATCCC 344 (GAA)12 63 [15] R: GGTGGTGATTGGACTGGTAGGAG L5 F: GCCTGTGTTTGGAGTATGGA 229 (AG)35 63 [15] R: TTGAGTGGAGGAAGAAGTAGAAG L6 F: GAGAAGGGCATCAACACCAAC 259 (CT)31 63 [15] R: GCCTCTCCTAAGCTTTACCCA L8 F: GTGCTCTCTCTTGATTGTTCG 237 (CT)32 63 [15] R: CGGATAGGGTGATATTGTGTG L11 F: AAGTCCGATCTCGCAAAC 283 (AG)34 63 [15] R: CTCTTACCCTTCTTCCACC L13 F: CGTCTTCGTTTCGCTACT 218 (GAA)10 63 [15] R: CCTTCTACTTCCCCAATCT L14 F: TCTCGCCATCCTACTTCG 432 (TTC)10 63 [15] R: GGTTTACGGTGACCTTCG L15 F: AGGTTCGTCGGAGTTAGG 333 (AG)33 63 [15] R: TTGCGTCAATGTTGCTTC L17 F: AACAGGAGAAGGGAAGCAATGG 375 (CTT)10 63 [15] R: GCCTTCAGCAATGGTGTCGG L18 F: CAAGGGTGCCATGGTAGTGATAA 260 (GA)36 63 [15] R: AGCCTGACCCACGCACCTATAC L21 F: AGTAATACCAGCAGTACCAGTC 126 (AGA)11 63 [15] R: CAGATAGCATCAGAAGCAGA L23 F: AGGAATTGGAGCCGTTGGTTGT 266 (TCT)12 60 [15] R: TACATTTGAGTCGGTTCCGGTTC L24 F: GTCACAGCCCCCAAAGAATA 100 (AGG)5 60 [14] R: GTTTCCCGCCATCACTCTTA L30 F: CCCCAAAATCACATTTCACC 218 (CCTTC)5 60 [14] R: TCAACAGTTGCTTGGAATCG Data analysis
-
Null alleles for each locus were detected using Micro-Checker 2.2.3[16]. The parameters of genetic diversity, including the observed number of alleles (Na), effective number of alleles (Ne), observed heterozygosity (Ho), expected heterozygosity (He), Shannon's information index (I), and gene flow (Nm) for different loci and populations, were calculated using GenALEx 6.5 software[17]. A Mantel test was used to assess the genetic distance between populations in relation to geographic distance and altitude difference. The correlation between genetic distance, geographic distance, and altitude difference was analyzed. Polymorphic information content (PIC) and fixation index (F) for different loci were calculated using Cervus software[18], in addition to performing a Hardy-Weinberg equilibrium test. Furthermore, HP-Rare software[19] was used to determine the allele richness (Ar) and private allele richness (PAr) of the populations. FSTAT software[20] was applied to determine Nei's genetic diversity index (H), genetic differentiation coefficient (Gst), and inbreeding coefficient (Fis) of populations. Arlequin software[21] was utilized to examine the molecular variance (AMOVA) within and between populations.
Structure software[22] was used to analyze the genetic structure of the population. First, the number of clusters was set between 2–9, and 20,000 iterations of Markov chain Monte Carlo (MCMC) were run for 10 repetitions with a burn-in period of 10,000 iterations. Next, Structure Selector[23] online (
https://lmme.ac.cn/StructureSelector/ ) was used to calculate the ΔK value, which will determine the optimal grouping number, K. The sampling analysis using CLUMPP software[24] was repeated to generate a population genetic structure diagram using Distruct software[25]. Additionally, individual evolutionary trees were built using genetic distance and the neighbor-joining method with MEGA 11 software[26]. -
The null allele frequency of each locus was less than 0.25 and the average was 0.07 (Table 3). Therefore, the null alleles had no significant effect on the estimation of population differentiation. A total of 150 alleles were detected in 460 samples using 16 loci, resulting in an average of 9.38 alleles per locus. The locus with the highest number of alleles was L15, with 55. Additionally, an average of 5.13 effective alleles per locus was observed, resulting in 82.1 effective alleles, and no ineffective alleles were found. Shannon's information index ranged from 0.93 to 2.26, averaging 1.67, suggesting high genetic diversity among the nine natural Fujian P. bournei populations at different loci. The average polymorphic information content was 0.86, all greater than 0.5, indicating high polymorphism at all 16 loci, effectively reflecting the genetic diversity of natural populations of P. bournei. The observed heterozygosity varied greatly across different loci, ranging from 0.32 to 0.99, with an average of 0.55, while the expected heterozygosity ranged from 0.50 to 0.84, with an average of 0.73. Except for loci L5 and L21, the expected heterozygosity at the other loci was higher than the observed heterozygosity, indicating a higher frequency of homozygotes in most loci. Furthermore, the average gene flow among all loci was 1.51, all greater than 1, indicating sufficient gene flow between populations. Four loci, namely L8, L13, L24, and L30 deviated from the Hardy-Weinberg equilibrium,while the remaining loci were in equilibrium.
Table 3. Genetic diversity parameters of P. bournei at different loci.
Locus Na Ne I Ho He PIC Gst Nm F Null HW L1 9 4.36 1.65 0.41 0.76 0.91 0.149 1.26 0.46 0.15 NS L2 7 4.80 1.74 0.46 0.78 0.84 0.088 2.25 0.42 0.00 NS L3 7 4.64 1.67 0.45 0.76 0.86 0.113 1.76 0.45 0.11 NS L5 8 6.37 2.05 0.99 0.83 0.94 0.106 1.96 −0.20 0.00 NS L6 17 9.27 2.26 0.57 0.84 0.95 0.109 1.84 0.37 0.00 NS L8 13 3.93 1.45 0.34 0.67 0.82 0.175 1.09 0.55 0.01 ** L11 9 7.08 2.02 0.68 0.81 0.94 0.134 1.49 0.16 0.10 NS L13 7 2.69 1.00 0.35 0.51 0.70 0.291 0.58 0.31 0.07 ** L14 11 6.32 1.88 0.47 0.80 0.90 0.108 1.84 0.45 0.09 NS L15 15 6.90 2.03 0.46 0.81 0.96 0.139 1.38 0.43 0.08 NS L17 7 4.67 1.63 0.63 0.76 0.84 0.096 1.99 0.17 0.12 NS L18 9 6.13 1.94 0.32 0.81 0.94 0.129 1.51 0.61 0.21 NS L21 8 3.83 1.38 0.81 0.67 0.80 0.179 1.10 −0.18 0.13 NS L23 9 5.69 1.83 0.70 0.80 0.88 0.095 2.15 0.16 0.00 NS L24 6 2.48 0.93 0.45 0.50 0.74 0.350 0.45 0.15 0.00 ** L30 8 2.98 1.18 0.64 0.64 0.70 0.133 1.52 0.01 0.00 * Total 150 82.14 Over all 9.38 5.13 1.67 0.55 0.73 0.86 0.15 1.51 0.27 0.07 Na: number of alleles, Ne: effective number of alleles, I: Shannon's information index, Ho: observed heterozygosity, He: expected heterozygosity, PIC: polymorphic information content, Gst: genetic differentiation coefficient, Nm: gene flow, F: fixation index, Null: null allele frequency, HW: Hardy-Weinberg equilibrium, NS: no significant, * p < 0.05 deviation from Hardy-Weinberg equilibrium, ** p < 0.01 extremely significant deviation from Hardy-Weinberg equilibrium. Genetic diversity of different populations
-
The overall level of genetic diversity in the nine natural populations of P. bournei was relatively high (Table 4). Still, there were significant differences in genetic diversity among the different populations. The Fujian Shunchang (FJSC) population had the highest level of genetic diversity (Na = 13.06, Ne = 7.48, Ho = 0.52, He = 0.83). On the other hand, the Jiangxi Chongyi (JXCY) population had the lowest genetic diversity level (Na = 7.50, Ne = 3.69, Ho = 0.31, He = 0.60), with the highest inbreeding coefficient (Fis = 0.50). The observed heterozygosity of each population was generally lower than the expected heterozygosity, and the inbreeding coefficient was greater than zero, with an average of 0.39, indicating the phenomenon of heterozygote deficiency and inbreeding in the population.
Table 4. Genetic diversity parameters of different natural populations of P. bournei.
Population Na Ne I Ho He Ar PAr Fis H FJSC 13.06 7.48 2.10 0.52 0.83 11.17 1.64 0.38 0.84 FJYX 10.13 6.68 1.93 0.44 0.81 9.21 0.80 0.46 0.82 GZTJ 13.06 6.73 2.01 0.41 0.80 11.35 1.29 0.49 0.81 FJJO 8.81 5.33 1.74 0.44 0.76 8.81 0.90 0.45 0.78 FJZH 8.56 4.83 1.65 0.52 0.76 7.53 1.10 0.32 0.77 FJMX 7.00 4.06 1.47 0.47 0.69 6.36 0.48 0.33 0.70 FJJY 8.06 4.24 1.55 0.64 0.71 7.27 0.74 0.11 0.72 GDLZ 4.69 3.16 1.21 0.38 0.65 4.43 0.14 0.43 0.66 JXCY 7.50 3.69 1.33 0.31 0.60 6.60 0.57 0.50 0.61 Mean 8.99 5.13 1.67 0.46 0.73 8.08 0.85 0.39 0.75 Na: number of alleles, Ne: effective number of alleles, I: Shannon's information index, Ho: observed heterozygosity, He: expected heterozygosity, Ar: allele richness, PAr: private allele richness, Fis: inbreeding coefficient, H: Nei's genetic diversity index. Genetic differentiation of populations
-
The results of the AMOVA analysis (Table 5) show that the genetic differentiation coefficient (FST = 0.162) between populations is greater than 0.15, indicating a high level of genetic differentiation among populations. Moreover, the intrapopulation variation (83.8%) is greater than the interpopulation variation (16.2%), suggesting that the genetic variation of P. bournei primarily exists within populations.
Table 5. Analysis of AMOVA molecular variance of natural populations of P. bournei.
Source of variation d.f. Sum of squares Variance components Percentage of variation p FST Among populations 8 984.88 1.15 16.20 < 0.01 0.162 Within populations 911 5425.84 5.96 83.80 Total 919 6410.72 7.11 The Mantel test results indicated a significant positive correlation (R2 = 0.18, p = 0.06) between genetic distance and altitude difference (Fig. 2b) between populations. However, there was no significant correlation with geographical distance (Fig. 2a), suggesting that altitude differences primarily influence the genetic differentiation of natural populations of P. bournei.
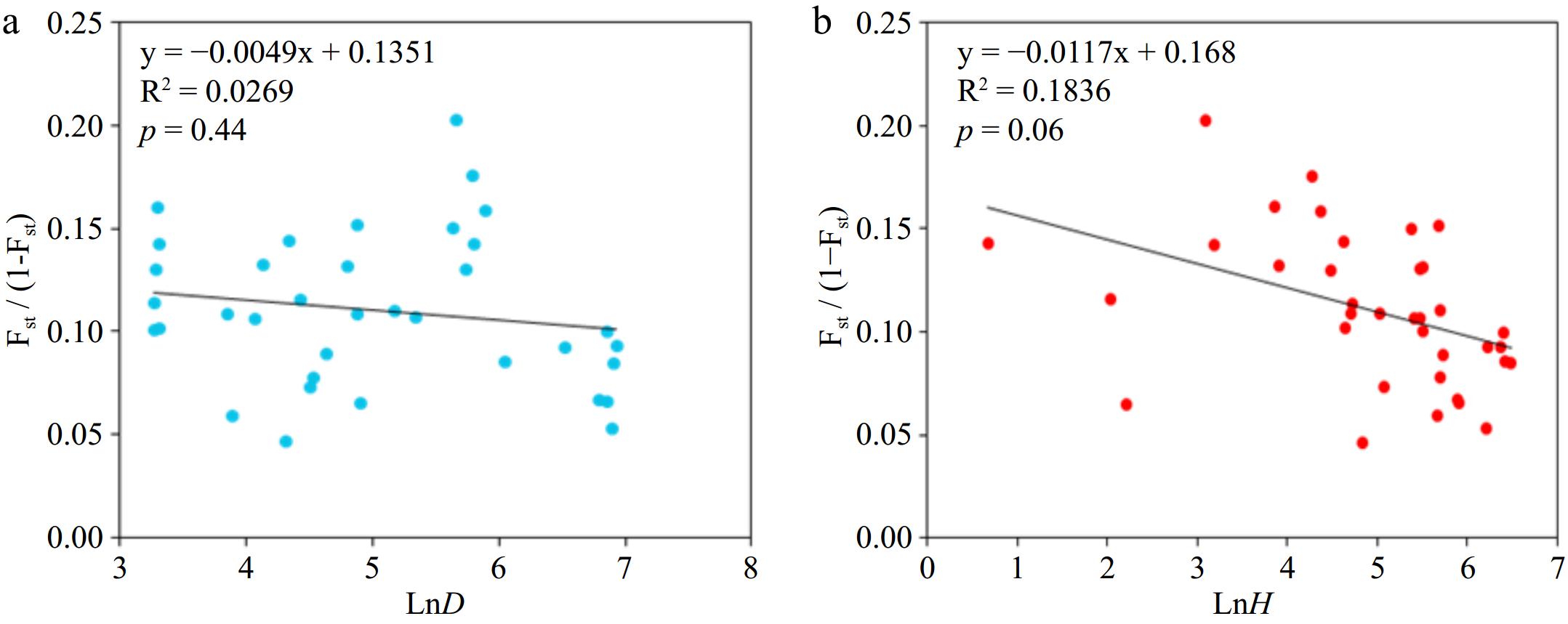
Figure 2.
Mantel test of genetic differentiation between (a) populations and geographical distance and (b) altitude difference.
Genetic distance and genetic consistency between populations
-
According to Fig. 3, the range of Nei's genetic distance and genetic consistency between populations was 0.47–1.48 and 0.23–0.63, respectively. These findings indicated some genetic differentiation among populations, which was consistent with the results of AMOVA. Despite their proximity, Fujian Zhenghe (FJZH) and Fujian Jianyang (FJJY) exhibited the largest genetic distance (1.48) and the smallest genetic consistency (0.23), suggesting a relatively distant relationship. In contrast, Guizhou Taijiang (GZTJ) and Jiangxi Chongyi (JXCY) had the smallest genetic distance (0.47) and the largest genetic consistency (0.63), implying a relatively close relationship.
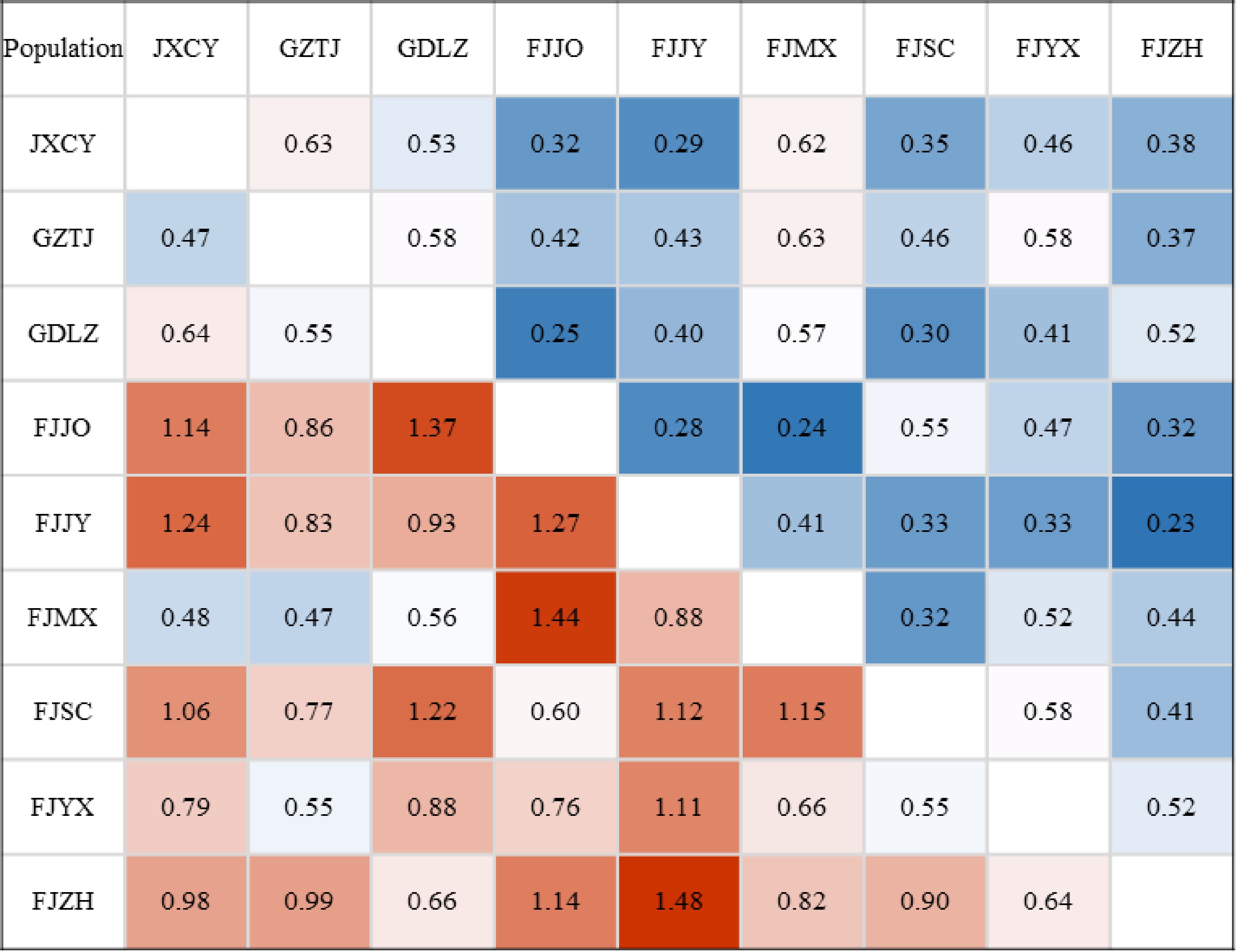
Figure 3.
Genetic distance and genetic consistency between populations. Note: Below the diagonal was genetic distance, above the diagonal was genetic consistency.
Genetic structure of populations
-
The results of the structure analysis indicated that the genetic differentiation of each population was most effectively reflected when K = 2 (Fig. 4). The maximum value of ΔK suggested that the genetic composition of all individuals could be classified into two clusters (Fig. 5). The first group (represented by the blue section) comprised Fujian Mingxi (100%), Guangdong Lianzhou (100%), Fujian Jianyang (98%), Jiangxi Chongyi (92.31%), and Guizhou Taijiang (54%). The second group (represented by the orange section) included Fujian Shunchang (100%), Fujian Jianou (100%), Fujian Youxi (81.82%), and Fujian Zhenghe (62%). In conclusion, the population of Fujian Mingxi, Guangdong Lianzhou, Fujian Shunchang and Fujian Jianou exhibited lower gene penetration, while other populations underwent varying degrees of gene exchange.
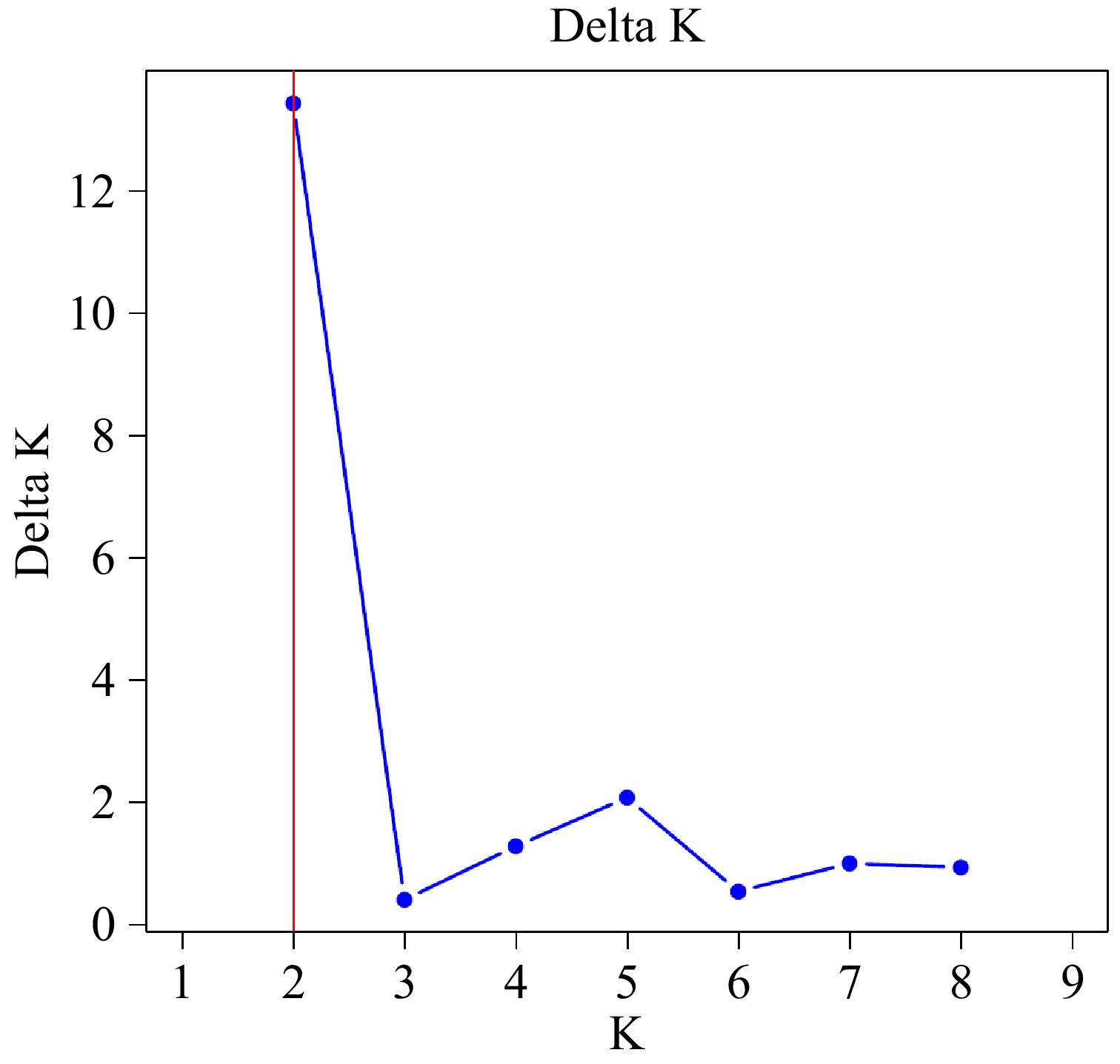
Figure 4.
The relationship between the optimal group number K and the inferred value ΔK.
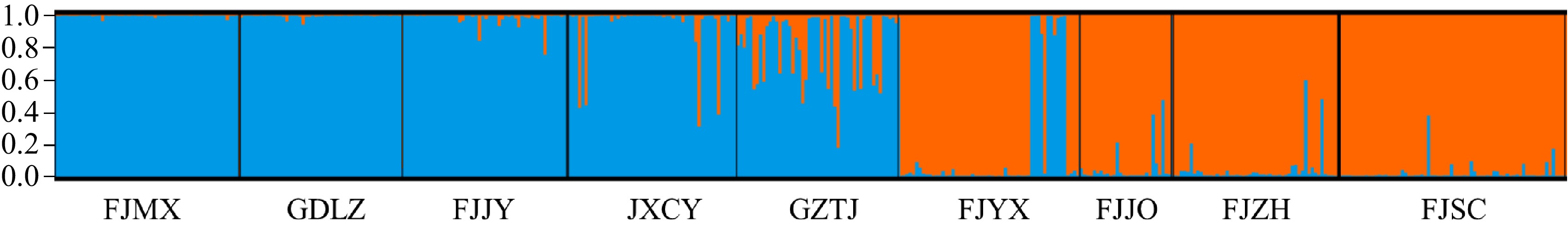
Figure 5.
Population genetic structure based on STRUCTURE analysis.
After analyzing the Q values for all individuals (Fig. 5), 421 individuals were discovered to have Q values greater than 0.6, accounting for 91.5% of the total. This indicated that the genetic composition of the individuals in these populations was relatively uniform, with only a few individuals exhibiting mixed genetic ancestry. On the other hand, the remaining 49 individuals had Q values less than 0.6, accounting for 8.48%. This suggests that the genetic composition of these individuals was more mixed.
The neighbor-joining method was used to construct a population clustering tree according to the genetic distance between individuals (Fig. 6). Based on this analysis, individuals from the tested populations were classified into two groups, consistent with the results obtained from the structure analysis. Group I consisted of 278 individuals, primarily sourced from individuals belonging to five distinct populations: Mingxi in Fujian, Jianyang in Fujian, Chongyi in Jiangxi, Lianzhou in Guangdong, and Taijiang in Guizhou. Furthermore, it included 10 individuals samples from Youxi in Fujian, eight individuals from Shunchang in Fujian, two individuals from Jianou in Fujian, and one individual from Zhenghe in Fujian. Group II contained 182 individuals, mainly from individuals of four populations including Zhenghe, Youxi, Jianou, and Shunchang in Fujian, as well as one individual from Taijiang, Guizhou.
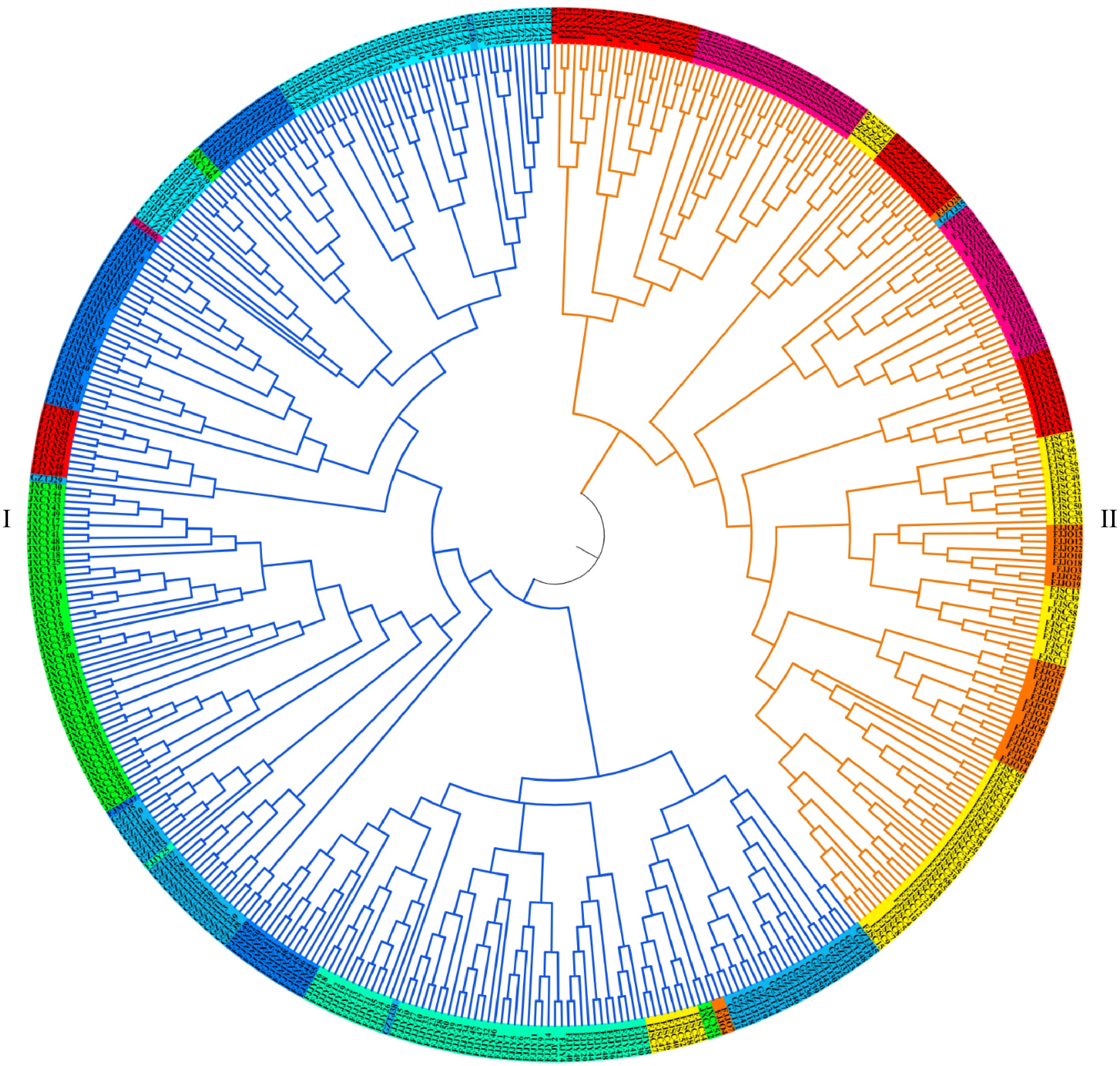
Figure 6.
Cluster analysis based on genetic distance among individuals.
-
The use of primers for polymorphic loci is a requirement when performing genetic diversity analyses. The level of polymorphism detected by the primers can be reflected in the polymorphism information content (PIC). If the PIC value is greater than 0.5, it indicates that the primers have a high contribution rate and adequate polymorphism and can fully portray the genetic diversity of populations[27]. Here, 16 primer pairs were used to examine the genetic diversity of 460 samples drawn from nine natural populations of P. bournei. The average PIC value obtained was 0.86. These results suggest that the SSR loci used in this study had an adequate polymorphism and were suitable for analyzing genetic diversity. They provided accurate and effective information on the genetic diversity of P. bournei populations in their natural habitats.
Genetic diversity within and between species is crucial for evolution and conservation[2]. Furthermore, a high level of genetic diversity within a species leads to great adaptability to environmental changes and improved vitality[28]. The current study investigated the genetic diversity of the natural population of P. bournei, which demonstrated the presence of 150 alleles, an expected heterozygosity range of 0.50 to 0.84, and an average value of 0.73. In particular, although the Fujian Jianou population had a small population size (28), it had a high level of expected heterozygosity (He = 0.76).
Furthermore, the genetic diversity of our studied species was found to be roughly equivalent to that of the relic species Ginkgo biloba (He = 0.808)[29], lower than the endangered species Semiliquidambar cathayensis (He = 0.816)[30] and the widely-distributed P. menziesii (He = 0.90)[11], significantly higher than that of the relic species G. pensilis (He = 0.272)[1]. Therefore, despite human disturbance, the natural population of P. bournei has maintained a high level of genetic diversity, indicating its potential for genetic evolution[31]. This may be attributed to the longer lifespan and shorter fragmentation duration of P. bournei, as the genetic diversity changes resulting from fragmentation have not yet been manifested[32].
Genetic diversity parameters can reveal the genetic structure and differentiation within a species. This study compared genetic parameters from natural populations of P. bournei from different geographical regions and found differences in genetic diversity among the different populations of the species. The population from Shunchang, Fujian, had the highest genetic diversity, while the population from Chongyi, Jiangxi, had the lowest. The Shunchang population covered a relatively large area with a high population density and large distances between individuals, growing in a scattered manner conducive to insect pollination and cross-pollination. Additionally, the Fujian Shunchang population was located at a high altitude (430 m), was relatively less affected by human activity, and had a lower rate of habitat fragmentation, maintaining its original population size and reducing the possibility of genetic drift, which maintained a high level of genetic diversity. Similarly, the population located at the highest altitude (800 m) in Taijiang, Guizhou, also had a high level of genetic diversity.
In contrast, the Chongyi, Jiangxi population had a small area with short pollination distances between individuals, leading to a greater likelihood of inbreeding and inbreeding depression. P. bournei also exhibited strong understory regeneration ability, and this population showed a phenomenon of parent-offspring coexistence, exhibiting prominent kinship relationships among individuals. Furthermore, the Jiangxi Chongyi populations were located at a low altitude, close to human settlements, and was heavily affected by human disturbances, resulting in small population sizes. The population at the lowest altitude in Lianzhou, Guangzhou, had a similar situation, with less genetic diversity. After conducting the sampling survey, it was discovered that although Zhenghe, Fujian, has the largest population area of the nine populations studied, the individuals within the population are highly concentrated, resulting in a high probability of inbreeding. This, in turn, has led to moderate genetic diversity within the population. Therefore, conservation and utilization strategies for P. bournei germplasm resources should prioritize populations with high genetic diversity, such as those in Shunchang, Fujian, which is well-preserved and has a large population size.
Perennial interspecific hybrids with a broad distribution and strong adaptability are believed to exhibit higher levels of population genetic differentiation[2]. Therefore, the population genetic differentiation coefficient is essential for evaluating the population genetic structure. A differentiation coefficient of 0.05–0.15 between populations is usually considered a moderate differentiation level. This study found that the natural population of P. bournei had a genetic differentiation coefficient of 0.162, which was at a high level. It was higher than that of endangered species Ormosia henryi (Gst = 0.0918)[33] and G. biloba (Gst = 0.093)[29] but lower than that of G. pensilis (Gst = 0.452)[1], and similar to that of Taxus mairei (Gst = 0.140)[34].
P. bournei is a woody perennial plant with many suitable habitats. The species has undergone long-term evolutionary sedimentation, resulting in rich genetic variation. However, due to overharvesting and human disturbances over a prolonged period, the area and number of natural populations have declined sharply, accelerating the process of habitat fragmentation. The reduction in population size may increase inbreeding and lead to population decline. However, since habitat fragmentation in natural populations of P. bournei is relatively recent, the degree of reduction in genetic differentiation levels is still relatively small. This study also found that in nine populations, about 64.8% of the individuals had a similar genetic composition. The observed heterozygosity was lower than expected, indicating a lack of sufficient heterozygotes, and the natural population of P. bournei had to inbreed. As an allogamous plant, habitat fragmentation restricts pollen dispersal, reducing the probability of random mating and making inbreeding more likely in the population. Therefore, a reasonable evaluation of the genetic structure of the natural population of P. bournei should also consider its genetic mating characteristics. Further research into the genetic mating system of P. bournei is needed to evaluate its genetic diversity more accurately and to provide a scientific and comprehensive theoretical basis for formulating appropriate protection strategies.
Molecular variance analysis revealed that genetic variation in P. bournei primarily occurred within populations, similar to outcrossing plants such as T. mairei and G. biloba. P. bournei reproduces through cross-pollination, and its pollen can travel long distances, promoting strong gene flow and genetic exchange between populations, leading to a gradual convergence of gene frequencies. Consequently, conservation efforts should prioritize genetic diversity within populations. In addition, altitude differences cause water, heat, light, and soil variations, leading to plant adaptation and genetic differentiation among populations[35,36]. This study found that genetic distance among P. bournei populations strongly correlates with altitude differences but is not significantly related to geographic distance, consistent with previous research on the woody plant Amygdalus mira in the tibet plateau in China[37]. This result suggests that natural selection in different environments at varying altitudes leads to significant genetic differences among populations. However, populations at the same altitude exhibit similar genetic variations.
The population genetic structure results of P. bournei indicated that there may be two gene sources in the tested materials. There was varying degrees of gene penetration between populations, and some individuals within the populations had complex genetic relationships. Although gene penetration between populations was not strictly clustered according to geography, it was still affected by some geographical isolation. Despite Taijiang in Guizhou, situated on the Yunnan-Guizhou Plateau, and Lianzhou in Guangdong, and Chongyi in Jiangxi, located in the Nanling Mountains, are situated far away from Jianyang and Mingxi in Fujian, which are near the Wuyi Mountains, these five natural populations all reside in high-altitude mountainous environments and may be subject to similar pressures of natural selection. In the long-term evolution process, there may be gene exchange between them, so they have relatively similar genetic structures. Fujian Zhenghe, Fujian Jianou, Fujian Shunchang, and Fujian Youxi are four populations situated in the low hilly regions of central and northern Fujian, sharing similar growth environments. Due to their proximity to each other, there may be seed exchange between these populations. The pronounced genetic difference between these two groups could potentially stem from geographical isolation, which is a result of the significant environmental differences between mountain ranges, plateaus, and hills.
-
Although P. bournei has been heavily harvested, the relatively high genetic diversity suggests that there is still immense potential for genetic evolution within the natural populations. Genetic diversity varied noticeably among different populations, with the Shunchang population in Fujian boasting the largest geographical area, scattered distribution of individuals, and the highest level of genetic diversity. Therefore, this population deserves priority protection. Despite the low genetic diversity in the Lianzhou, Guangdong, and Chongyi, Jiangxi populations, the individuals in these groups still contained many alleles. Therefore, it is crucial to prevent the loss of genetic diversity due to human activities or natural disasters. Genetic variation in P. bournei existed mainly within populations, and genetic differentiation between populations was moderate. The nine natural populations of P. bournei could be divided into two categories, with varying degrees of gene infiltration between populations. Group I included populations in Fujian Mingxi, Jianyang Fujian, Jiangxi Chongyi, Lianzhou Guangdong, and Guizhou Taijiang. Group II consisted populations in Zhenghe, Youxi, Jianou, and Shunchang from Fujian.
This work was supported by the Seed Industry Innovation and Industrialization Engineering Project of Fujian Province (ZYCX-LY-2021005); the Forestry Seedling Technology Research Project Phase VII of Fujian Province (ZMGG-0708).
-
The authors confirm contribution to the paper as follows: study conception and design: Fan H, Zhou Z; conducting the experiments: Wang Y; data curation: Wang Y; writing – original draft preparation: Wang Y; resources collection: Fan H, Pan X, Tang X; writing – review & editing: Zhou Z; project administration and supervision: Fan H, Zhou Z. All authors reviewed the results and approved the final version of the manuscript.
-
In this manuscript, no raw sequences, including nucleic acid and protein sequences, are presented. However, all calculation data are held by the authors and can be provided to reviewers or submitted to any database upon request from the corresponding author.
-
The authors declare that they have no conflict of interest.
-
Received 8 July 2024; Accepted 8 October 2024; Published online 3 December 2024
-
There was a relatively high level of genetic diversity.
High levels of differentiation existed between populations.
The genetic distance among populations was positively correlated with altitude.
The nine natural populations were classified into two categories.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wang Y, Fan H, Zhou Z, Pan X, Tang X. 2024. Genetic diversity and genetic structure of the natural population in the critical production area of Phoebe bournei. Tropical Plants 3: e040 doi: 10.48130/tp-0024-0039 

Genetic diversity and genetic structure of the natural population in the critical production area of Phoebe bournei
- Received: 08 July 2024
- Revised: 27 September 2024
- Accepted: 08 October 2024
- Published online: 03 December 2024
Abstract: Phoebe bournei, also known as 'nanmu', is renowned for its exceptional wood quality. However, the natural forest resources of P. bournei are increasingly scarce due to excessive logging and usage. This study utilized EST-SSR molecular marker technology to assess the genetic diversity and structure of nine natural populations of P. bournei, providing a theoretical foundation for the conservation and use of its germplasm resources. There was a notably high level of genetic diversity, with an average expected heterozygosity of 0.73. There were significant variances in genetic diversity among the populations. The Shunchang population in Fujian Province (China) exhibited the highest genetic diversity (He = 0.83), which should be prioritized for conservation due to its extensive area and dispersed individual distribution. The population in Chongyi, Jiangxi Province (China), had the lowest genetic diversity (He = 0.60) due to inbreeding and its relatively small area. Genetic differentiation occurred primarily within populations (83.8%). However, high levels of differentiation existed between populations (FST = 0.162). The genetic distance between populations was positively correlated with altitude, suggesting that altitude may impact the genetic differentiation of natural populations of P. bournei. Genetic structure and cluster analysis revealed that the nine natural populations of P. bournei were classified into two categories.
-
Key words:
- Phoebe bournei /
- EST-SSR /
- Genetic diversity /
- Genetic structure /
- Natural population