









-
Establishing a high-frequency regeneration system is a prerequisite for genetic transformation, which is one of the most effective biotechnological approaches in crops[1,2]. Strawberry (Fragaria spp.), an economically important berry crop, has become a model crop for fundamental research in recent years[3], and numerous studies have been undertaken to develop regeneration systems of strawberry cultivars or species, including the octoploid genotype LF9[4], Chandler[5] and the diploid wild species Fragaria vesca[6]. The diploid wild Chinese strawberry species Fragaria nilgerrensis Schlechtendal ex J. Gay is mainly distributed in Sichuan, Yunnan, Guizhou, Shaanxi, Hubei, Chongqing, and Hunan Provinces (China) with many excellent characteristics, such as fruit with white color, peach fragrance, and strong disease resistance[7]. Thus, the regeneration of F. nilgerrensis has attracted wide research attention[8]. To date, a few studies on regeneration in F. nilgerrensis have been reported. For example, the shoot regeneration from detached leaves was established, and the regeneration frequency did not reach 80%[9]. Although the callus was induced from F. nilgerrensis leaves with an induction rate of 95%, the differentiation of callus into plantlets did not continue to be studied[10]. Despite reports of regeneration in F. nilgerrensis, the regeneration efficiency remains low, thereby preventing its application to genetic transformation. Therefore, there is an urgent need to establish a high-efficiency regeneration system for F. nilgerrensis.
With the development of genomics of strawberries, the understanding of gene function is gradually deepened. Both gene manipulation technologies including overexpression and knockout/down and gene editing technology (CRISPR/Cas9) depend on a high-efficiency transformation system[11−13]. A large number of studies on gene manipulation and trait improvement through Agrobacterium-mediated transformation have been carried out in different strawberry species[14−17]. Pi et al. generated diploid strawberry F. vesca variety 'H4' transgenic lines with parthenocarpic fruit growth and delayed fruit ripening by mutating gene FveSEP3 generated by CRISPR/Cas9[18]. The RAP gene was successfully transformed into the F. vesca variety 'Yellow Wonder' and octoploid cultivated strawberry respectively by overexpressing and knockout technology. The results showed that RAP is one important gene in the fruit color breeding of strawberry[19]. It is reported that strawberry cultivars with a high regeneration rate are more likely to regenerate into plantlets during the Agrobacterium-mediated genetic transformation[16]. For example, a high-efficiency regeneration method was developed with 6.67 shoots per explant in the strawberry cultivar 'Chandler'. Based on this, the genetic transformation efficiency was up to 31.25%[5,20]. Therefore, establishing a stable and high-efficiency transformation system that relies on efficient regeneration is essential for strawberry. F. nilgerrensis, an important diploid strawberry, could be used as a model plant species for various fundamental research due to its relatively small genome size (250 Mbp). The completion of sequencing and assembling of F. nilgerrensis has accelerated the identification and mining of its candidate genes controlling some traits[21]. However, the lack of an efficient genetic transformation system for this species has hindered the verification of these gene functions.
Various factors could influence strawberry regeneration and genetic transformation, including genotype, explant type, hormone concentration, Agrobacterium strain, infection time, antibiotic concentration, and so on. It has been reported that regeneration and genetic transformation efficiency are closely related to genetic background and the genotype is a major factor[13]. For example, the octoploid strawberry cultivars 'Festival' and 'Fortuna' have a higher percentage of callus induction than 'Sweet Charlie'[22]. The transformation percentage achieved in 'Rapella' was only 0.02%[14], while that in 'Hecker' was 68%[15]. These indicated wide differences in the regeneration and transformation rates among strawberry cultivars. The hormone is another crucial factor involved in the regulation of regeneration. The cytokinin thidiazuron (TDZ) and 6-benzylaminopurine (6-BA), and the auxins indole-3-acetic acid (IAA), 1-naphthlcetic acid (NAA), and 2,4-dichlorophenoxyacetic acid (2,4-D) are generally used for strawberry regeneration[10,23,24]. In particular, TDZ has been frequently utilized in many strawberry species for rapid plantlet regeneration, probably due to its function as a regulator of endogenous auxin levels and its cytokinin-like activity[5,25]. Other regeneration and transformation factors have also been studied in strawberries including exogenous nutrient casein hydrolysate, Fe-EDTA or KNO3[26−27], dark culture time[28], explant (leaf disc)[26], Agrobacterium strain, infection time, antibiotic types and concentrations[5]. The above factors were mainly studied in octoploid strawberry cultivars, however, the high ploidy and heterozygosity of this species pose problems and difficulties for further genetic and molecular studies[5,29]. Studies have shown that the diploid species with a relatively small genome size is a valuable resource for various fundamental research as model plant species[21,30]. However, only a few factors influencing regeneration and transformation have been studied in diploid F. nilgerrensis.
In the present study, several factors affecting in vitro regeneration of F. nilgerrensis leaf discs were optimized, including dark culture time, CH, TDZ/6-BA, and IBA concentrations to establish the efficient regeneration system that could guarantee the generation of transgenic shoots. Meanwhile, we analyzed callus morphology using paraffin sections to determine the regeneration pathways. Subsequently, the transgenic plants were obtained in Agrobacterium-mediated genetic transformation. PCR-positive transformants were confirmed by PCR and histochemical GUS, and transgene integration was identified by transformants regeneration. This study may be useful in gene functional research and trait improvement for F. nilgerrensis.
-
The effects of plant growth regulators (PGRs) on shoot differentiation were analyzed using different concentrations of TDZ (1, 2, 3, and 4 mg·L−1) and 6-BA (1, 2, 3, and 4 mg·L−1) combined with IBA (0.1, 0.2, and 0.3 mg·L−1). The analysis demonstrated that the rates of callus induction and regeneration and the number of regenerated shoots per explant varied significantly between the treatments at 45 d of inoculation (Table 1). At constant IBA concentration, the rate of shoot regeneration first increased and then decreased as TDZ concentration increased, which is consistent with the 6-BA treatment. The higher rates of callus induction and regeneration and more regenerated shoots per explant were obtained with 2 mg·L−1 TDZ. Similarly, when the concentration of TDZ or 6-BA was maintained constant, the three indexes reached the maximum at 0.2 mg·L−1 IBA. Simultaneously, lower callus formation and shoot regeneration rates were obtained when 6-BA was used instead of TDZ (Table 1). At 25 d of inoculation, most leaves curled, but the callus did not cover the entire leaf in the presence of 6-BA in the medium. In contrast, in the medium containing TDZ, thick, loose, yellow, and granular callus covered the whole leaves (Fig. 1a). The effect of TDZ on regeneration and callus induction was more obvious than that of 6-BA. In summary, the A2C2 medium containing 0.2 mg·L−1 IBA and 2 mg·L−1 TDZ exhibited the highest rates of callus induction (97.8%) and shoot regeneration (97.3%) and maximum regenerated shoots per explant (2.52) at 45 d of culture and was further used for genetic transformation.
Table 1. Effect of different PGRs on regeneration of F. nilgerrensis leaf discs at 45 d of culture.
Media code Concentration (mg·L−1) Callus induction rate (%) Shoot regeneration rate (%) Shoot no. per explant TDZ BA IBA A1C1 1 0.1 21.1 ± 1.91 i 9.1 ± 2.08 j 0.11 ± 0.04 gh A1C2 1 0.2 94.2 ± 2.14 ab 37.8 ± 1.91 e 0.40 ± 0.09 e A1C3 1 0.3 61.1 ± 1.91 e 0.0 ± 0.00 l 0.0 ± 0.00 i A2C1 2 0.1 80.0 ± 2.00 c 79.0 ± 3.60 c 1.47 ± 0.08 c A2C2 2 0.2 97.8 ± 0.57 a 97.3 ± 0.58 a 2.52 ± 0.09 a A2C3 2 0.3 64.5 ± 3.87 de 4.4 ± 1.93 k 0.07 ± 0.03 hi A3C1 3 0.1 25.6 ± 1.96 h 17.8 ± 1.91 h 0.19 ± 0.02 fg A3C2 3 0.2 92.1 ± 1.82b 89.5 ± 0.50 b 1.96 ± 0.07 b A3C3 3 0.3 62.2 ± 3.89 e 20.3 ± 0.58 g 0.25 ± 0.09 f A4C1 4 0.1 67.8 ± 1.91 d 0.0 ± 0.00 l 0.0 ± 0.00 i A4C2 4 0.2 45.6 ± 1.96 g 0.0 ± 0.00 l 0.0 ± 0.00 i A4C3 4 0.3 7.1 ± 2.54 j 0.0 ± 0.00 l 0.0 ± 0.00 i B1C1 1 0.1 5.6 ± 1.93 j 0.0 ± 0.00 l 0.0 ± 0.00 i B1C2 1 0.2 5.6 ± 1.93 j 0.0 ± 0.00 l 0.0 ± 0.00 i B1C3 1 0.3 27.8 ± 1.91 h 0.0 ± 0.00 l 0.0 ± 0.00 i B2C1 2 0.1 27.8 ± 1.91 h 0.0 ± 0.00 l 0.0 ± 0.00 i B2C2 2 0.2 47.7 ± 1.31 fg 14.3 ± 2.31 i 0.13 ± 0.05 gh B2C3 2 0.3 67.8 ± 1.91 d 66.7 ± 3.51 d 0.6 ± 0.10 d B3C1 3 0.1 25.2 ± 2.54 h 0.0 ± 0.00 l 0.0 ± 0.00 i B3C2 3 0.2 4.5 ± 1.91 j 0.0 ± 0.00 l 0.0 ± 0.00 i B3C3 3 0.3 50 ± 3.30 f 34.4 ± 1.96 f 0.35 ± 0.08 e B4C1 4 0.1 5.6 ± 1.91 j 0.0 ± 0.00 l 0.0 ± 0.00 i B4C2 4 0.2 8.9 ± 5.06 j 0.0 ± 0.00 l 0.0 ± 0.00 i B4C3 4 0.3 8.3 ± 1.67 j 0.0 ± 0.00 l 0.0 ± 0.00 i Values represent mean ± SE; Different lowercase letters indicate significant differences among treatments according to the LSD test (P < 0.05). 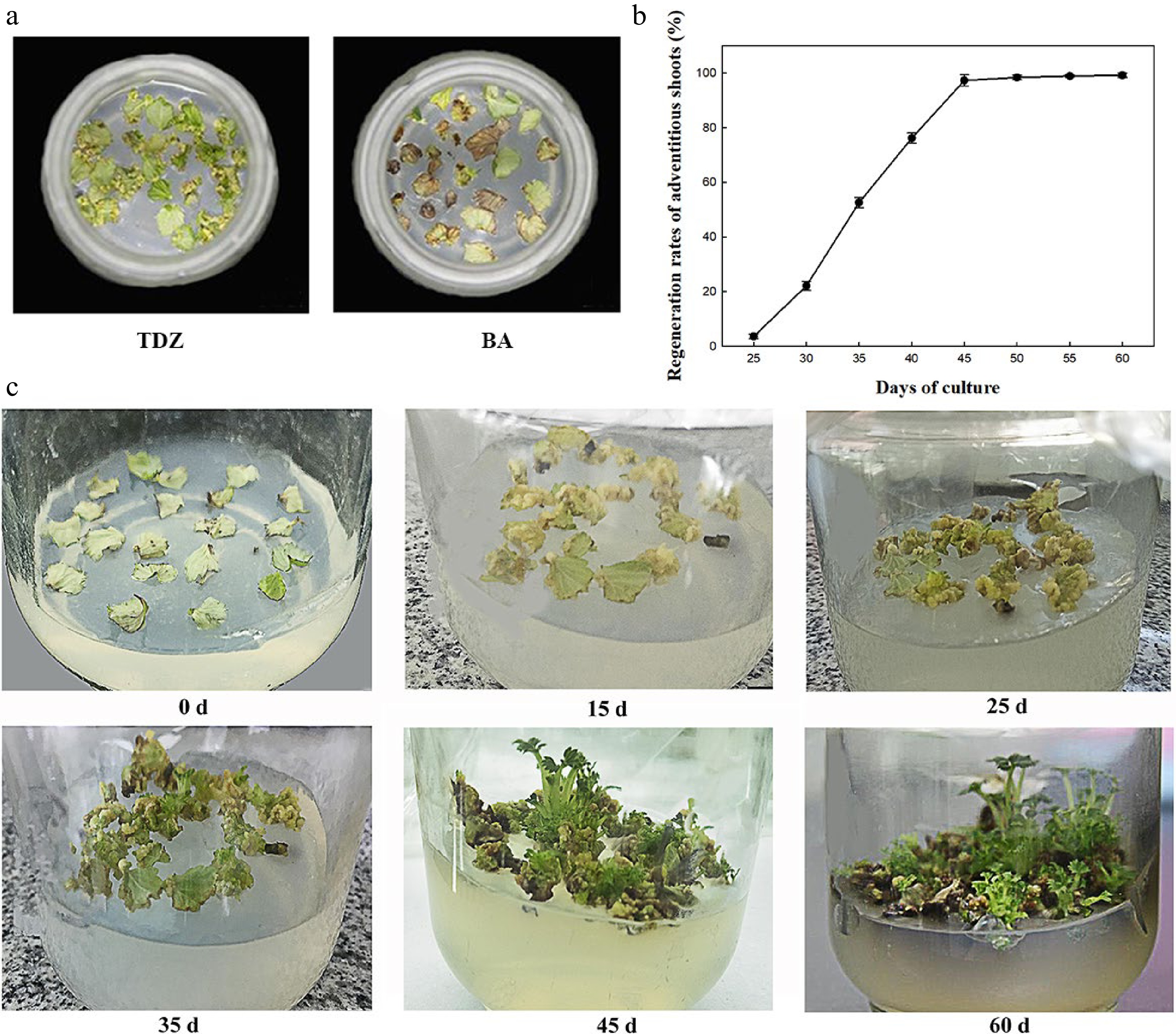
Figure 1.
Effect of PGRs on regeneration of F. nilgerrensis leaf discs. (a) Effects of TDZ and 6-BA on callus formation at 25 d of culture. (b) Shoot regeneration curve cultured on A2C2 medium for different days. (c) Shoot regeneration process cultured on A2C2 medium for different days.
The shoot regeneration rate was measured every 5 d on A2C2 medium (Fig. 1b). It can be seen from Fig. 1b that shoot regeneration rate was 2.6% at about 25 d of culture and the regeneration rate rapidly increased from 35 to 45 d of culture, reached the peak of 97.3% at 45 d of culture and became stable. Moreover, the process of callus induction and shoot regeneration were continuously observed on A2C2 media for 60 d (Fig. 1c). A large amount of callus was formed around 15 d of culture, and began to differentiate into shoots at 25 d of culture. The height of shoots reached 1.0−2.0 cm at 45−60 d of culture.
To analyze the influence of dark culture on regeneration efficiency, five treatments of dark culture with 0, 7, 14, 21, and 28 d respectively were used in the experiment. The callus weight and shoot regeneration rate first increased and then decreased as the dark culture duration increased (Fig. 2a), and at 45 d of inoculation, the shoot regeneration rate of 14 d dark culture was the highest (98.5%), which was similar to that of 21 d dark culture. Furthermore, the rate of callus induction and shoot regeneration rate were very low in the treatment of 0 d dark culture (Fig. 2c), which is significantly lower than that of 7 d dark culture. While dark culture for over 28 d led to browning and blackening of callus and decreased the callus induction and regeneration rates (Fig. 2c). These results indicated that dark culture duration affected the rate of callus induction and quality of callus, and then affected the regeneration rate.

Figure 2.
Effects of CH concentrations and dark culture duration on the regeneration of F. nilgerrensis leaf discs at 45 d of culture. (a) The callus weight and shoot regeneration rates under different CH concentrations and dark culture duration. (b) The callus growth curve on the media containing 0.0 and 0.5 g·L−1 CH. (c) Effect of dark culture for 0, 7, 14, 21, and 28 d on regeneration. (d) The effect of 0.0, 0.2, 0.5 , 0.7, and 1.0 g·L−1 CH on regeneration. Bars indicate the S.E. of the means; different lowercase letters indicate significant differences among the treatments according to the LSD test (P < 0.05).
The effects of different concentrations of CH on regeneration were evaluated in our study. After 45 d of culture, CH at five concentrations significantly affected the rates of shoot regeneration. The shoot regeneration rate and callus weight first increased and then decreased as CH concentration increased, and the highest regeneration rate (96.6%) and callus weight (7.2 g) was attained at 0.5 g·L−1 CH, similar to at 0.2 g·L−1 CH (Fig. 2a). The percentage of callus with thick, yellow, granular, and tight texture was higher at 0.2 or 0.5 g·L−1 CH, while the percentage of callus with brown color increased on the medium with 0.0, 0.7, or 1.0 g·L−1 CH (Fig. 2d).
In the current experiment, two types of callus appeared: Type I callus, the callus was light yellow and compact, with a higher regeneration rate; Type II callus, the callus was brown, massive, or spongy, with a lower regeneration rate. The effect of different concentrations of CH on the percentages of Type I and Type II callus were compared (Table 2). The induction rate of Type I callus was only 6.11% on the medium without CH. However, the highest induction rate of Type I callus (87.23%), and the lowest induction rate of Type II callus (9.44%) were obtained when the 0.5 g·L−1 CH was added to the medium. When CH concentration continued to increase, the induction rate of Type I callus began to decrease. Therefore, the addition of CH with optimal concentration in the medium was beneficial to the formation of Type I callus and the optimal concentration of CH was 0.5 g·L−1.
Table 2. The rates of Type I and II callus on the medium with different CH concentrations of F. nilgerrensis leaf discs at 45 d of culture.
CH concentration (g·L−1) No. of leaf discs Rate of callus induction (%) Rate of Type I callus (%) Rate of Type II callus (%) 0.0 60 75.56 ± 0.96 c 6.11 ± 0.96 e 69.44 ± 0.96 a 0.2 60 91.11 ± 0.96 b 81.11 ± 0.96 b 10.00 ± 1.67 d 0.5 60 96.67 ± 1.67 a 87.23 ± 1.93 a 9.44 ± 1.93 d 0.7 60 71.11 ± 1.92 d 21.67 ± 1.67 c 49.45 ± 2.54 b 1.0 60 62.78 ± 0.96 e 17.22 ± 0.96 d 45.56 ± 1.93 c Values represent mean ± SE; Different lowercase letters indicate significant differences among treatments according to the LSD test ( P < 0.05). Furthermore, the effects of 0.0 and 0.5 g·L−1 CH on the callus growth were compared every 5 d for 45 d. The growth curve presented a typical 'S' shape on both concentrations (Fig. 2b), but the callus growth was significantly higher at 0.5 g·L−1 CH than at 0.0 g·L−1 from 25 d to 45 d of culture. The biomass of the callus steadily rose with prolonged culture. The callus growth rate was the highest at 25 d to 35 d and became stable after 35 d.
Observation of callus paraffin sections
-
The process of callus growth in our experiment was as follows: first, the leaves began to curl, and the callus appeared within 7 d of culture when the leaf discs were cultured on the regeneration medium in the dark. A large amount of white callus was formed around 10−20 d of culture and covered with the whole leaves. Then some white callus turned into Type I callus at 20−35 d of culture on the A2C2 medium with 2.0 mg·L−1 TDZ, 0.2 mg·L−1 IBA, and 0.2 or 0.5 g·L−1 CH under the dark culture for 14−21 d of culture, and began to differentiate into shoots at 25 d of culture, which proved the regenerative pathway of F. nilgerrensis leaf discs was indirect organogenesis. However, the white callus turned brown (Type II) with a lower differentiation rate.
Light yellow and brown callus was observed at 25 d of culture through paraffin sections. Yellow callus was formed by smaller cells with regular shape, close arrangement, and obvious nuclei, which showed the morphological characteristics of embryogenic callus (Fig. 3a). However, the cells of brown non-embryonic callus were bigger, irregular, more loosely arranged, and highly vacuolated ones with larger intercellular spaces, and non-obvious nucleus (Fig. 3b). These results indicated that the regeneration pathway of indirect organogenesis for F. nilgerrensis leaf discs was yellow embryogenic callus as the intermediate process.

Figure 3.
Paraffin section observation of callus obtained from F. nilgerrensis leaf discs. (a) Type I callus (left) and its cells (right, black arrow), bar = 100 μm. (b) Type II callus (left) and its cells (right, black arrow), bar = 100 μm.
Optimization of Kan concentrations and Agrobacterium-mediated method for the transformation of F. nilgerrensis leaf discs
-
The non-transformed explants were cultured in the shoot regeneration medium supplemented with different Kan concentrations (0, 5, 10, and 15 mg·L−1) for screening of threshold concentrations and to inhibit non-transgenic shoots. The results showed that shoot differentiation was inhibited at 5 mg·L−1 Kan concentration and all explants died at 10 and 15 mg·L−1 Kan concentrations (Table 3). Therefore, 10 mg·L−1 Kan was selected as the threshold for non-transformed explants.
Table 3. The regeneration rates in different Kan concentrations of F. nilgerrensis leaf discs at 45 d of culture.
Kan concentration (mg·L−1) No. of leaf discs Rate of regeneration (%) 0 60 98.32 ± 2.01a 5 60 51.36 ± 1.62b 10 60 0.08 ± 0.01c 15 60 0.00c Values represent mean ± SE; Different lowercase letters indicate significant differences among treatments according to the LSD test (P < 0.05). Kan concentrations were set to 10, 20, 30, 40 and 50 mg·L−1 to determine the effects on transformation efficiency. The results showed that significant differences were detected among the different concentrations and the percentages of resistant shoot regeneration and transformation decreased with increasing Kan concentrations (Table 4). At 10 mg·L−1 Kan concentrations, the number of PCR-positive transformants was less than half that of resistant shoots, indicating some false positive plantlets. At 50 mg·L−1 Kan, the percentage of the resistant shoot regeneration and transformation efficiency was 0.00%, demonstrating a substantial impairment in the ability of shoot regeneration from leaf discs. Furthermore, we found that compared with 30 mg·L−1 Kan, although 90% resistant shoots were identified as PCR-positive transgenic lines using 20 mg·L−1 Kan, the resistant shoot regeneration rate and transformation percentage were greater, and a relatively high transformation efficiency was obtained. Therefore, 20 mg·L−1 Kan was used as the optimal concentration for efficient genetic transformation.
Table 4. Effect of Kan concentrations on Agrobacterium-mediated transformation of F. nilgerrensis leaf discs.
Kan concentration
(mg·L−1)No. of infected explants Rate of resistant shoot regeneration (%)
at 65 d post-infectionPercentage of transformation (%)
at 130 d post-infection10 60 24.74 ± 0.62a 10.98 ± 0.13a 20 60 10.25 ± 0.18b 9.22 ± 0.20b 30 60 3.33 ± 0.14c 3.22 ± 0.25c 40 60 1.67 ± 0.01d 1.67 ± 0.01d 50 60 0.00e 0.00e Values represent mean ± SE; Different lowercase letters indicate significant differences among treatments according to the LSD test (P < 0.05). The result of Agrobacterium-mediated transformation for 600 leaf discs of F. nilgerrensis showed the formation of callus at 15 d and the regeneration of resistant shoots at 65 d post-infection (Fig. 4). The putative transgenic plantlets were obtained through 60−90 d of elongation culture of resistant shoots. Meanwhile, the experiment showed the regeneration rates of resistant shoots (%) at 65 d post-infection for the three replicates were 10.12%, 9.56%, and 9.11% respectively, and the average value was 9.60%, which indicated that a relatively stable transformation system was established. PCR analysis of resistant transgenic lines will be performed to identify positive transgenic lines at 130 d post-infection.
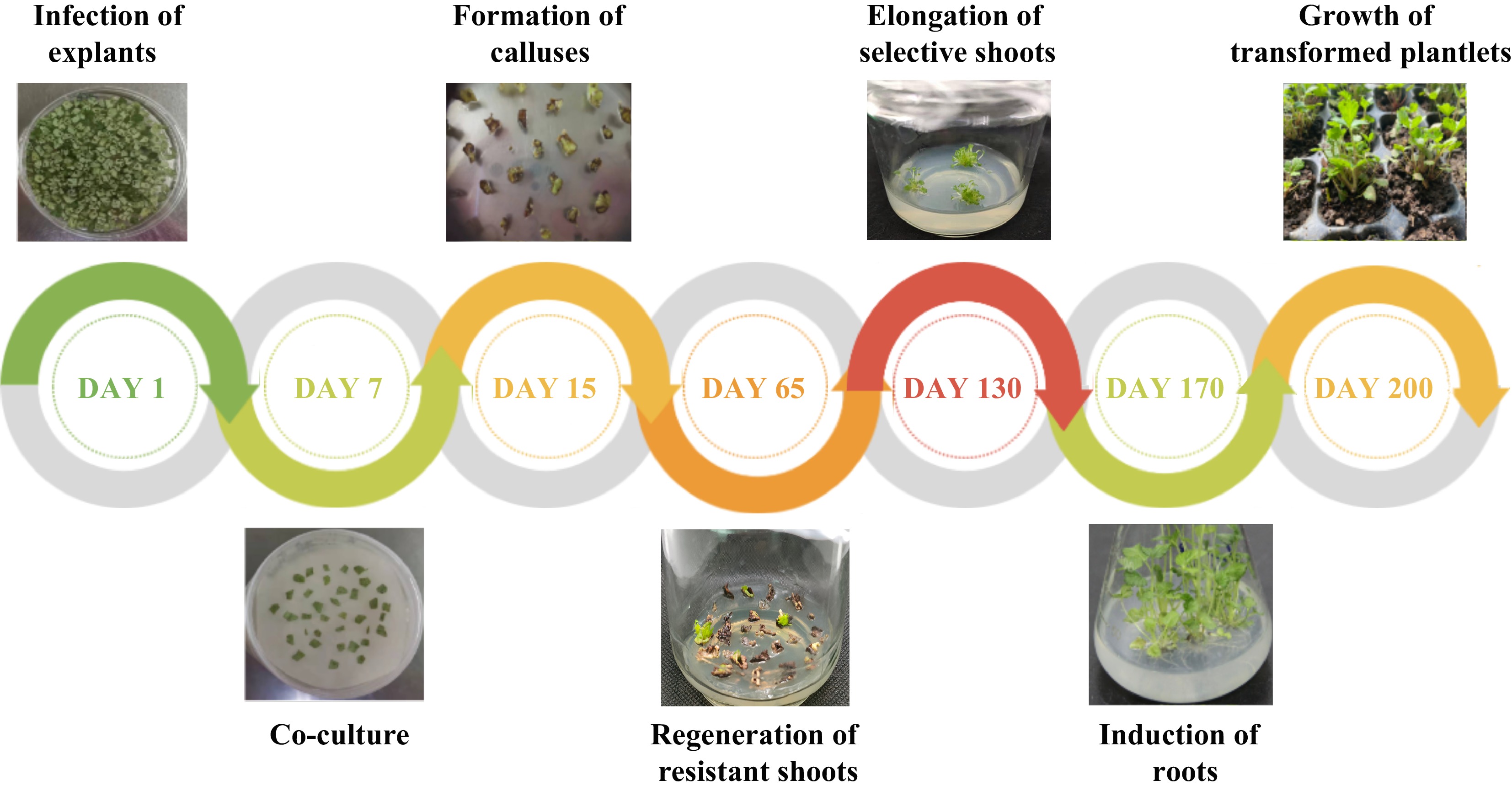
Figure 4.
Agrobacterium-mediated genetic transformation of F. nilgerrensis leaf discs.
Identification of transgenic lines
-
A total of 58 putative transgenic lines were recovered by Agrobacterium-mediated transformation of F. nilgerrensis leaf discs, of which 52 (89.66%) were confirmed as transgenic lines after PCR analysis. It can be seen from the PCR results that the 1,800 bp GUS gene was amplified to varying degrees from 52 positive transgenic lines (Fig. 5), while the negative controls showed no amplification. From the above, the transformation percentage for F. nilgerrensis leaf discs was 8.67% (52 out of 600). Meanwhile, using this system, it took approximately 4−5 months to form PCR-positive transgenic lines.
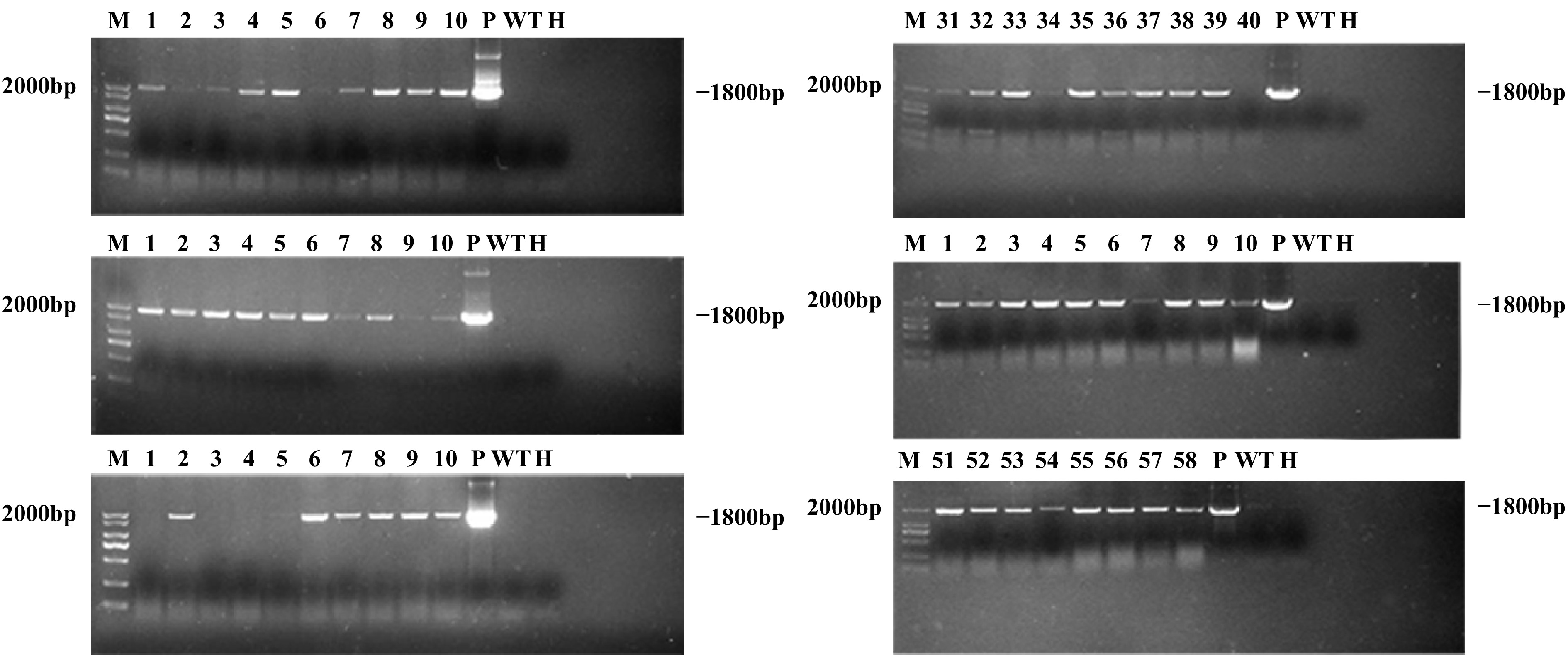
Figure 5.
PCR analysis of GUS gene in transgenic lines. Lane M, DNA marker; Lanes 1–58, 58 putative transgenic lines; Lane WT, wild-type plant; Lane P, plasmid control; Lane H, ddH2O.
GUS staining showed that the positive lines' whole or part of the leaves showed a blue reaction, which confirmed the expression of the GUS gene. Meanwhile, the control leaves displayed no blue GUS staining (Fig. 6).

Figure 6.
Histochemical GUS staining of transgenic lines. (a) Wild-type plant. (b)–(d) PCR-positive transgenic plants.
The regeneration rates of leaf discs from transgenic lines were assessed to further segregate Kan resistance transformants and identify the transgene integration. The results showed that the regeneration rates of leaf discs for three repetitions were 90%, 80% and 90%, respectively, with an average regeneration rate of 86.67%, which indicated the transgenic lines were resistant to Kan and the transgene integration of PCR-positive transformants (Fig. 7). Meanwhile, all leaf discs of the non-transgenic plants were brown within 45 d of culture.

Figure 7.
Transformants regeneration on medium with Kan. (a) Leaf regeneration of PCR-positive transgenic lines under Kan selective pressure. (b) Non-transformed plants (control).
-
The construction of a high-efficiency regeneration system is essential for the successful genetic transformation of strawberries. There are two ways to establish a strawberry regeneration system: organogenesis and somatic embryogenesis, both of which include direct and indirect regeneration. The indirect regeneration pathway is the formation of the callus as the intermediate process. Despite reports of somatic embryogenesis regeneration from various strawberry explants, this pathway was still much less efficient and generally occurred directly, without the formation of callus[5,32−34], which hinders genetic transformation. Therefore, the main regeneration pathway for strawberry is organogenesis. Numerous studies have demonstrated that the regeneration of various strawberry cultivars is based on the callus stage[9] and the formation of embryogenic callus with a crisp texture, compact structure, and nodular or tumor-like protrusions can significantly increase the regeneration rate[35]. The most effective induction rate (71.4%) of embryogenic callus was obtained for 'Jukhyan' strawberry anther after optimizing the factors including AgNO3, Fe-EDTA and types of media, and the regeneration rate was highest (92.7%) on 1/2 MS medium[36]. It is reported that adding yeast extract[37], CH[38], dicamba[39], AgNO3[40], carbon sources[41], and activated charcoal[35] into the medium increased the induction rate of embryogenic callus and affected the maintenance and differentiation rate of the embryogenic callus. For example, nutrients including CH and potassium nitrate that were added to the medium increased the rate of the callus and shoot formation for 'Allstar' strawberry leaf[42]. Some researchers showed that different CH concentrations affected the callus induction and status and identified 0.2−1.0 g·L−1 as the suitable concentration for the regeneration of different crops[37,43]. In our experiment, in the presence of CH at 0.2 or 0.5 g·L−1, embryogenic callus was formed at 20−35 d of culture and directly differentiated into shoots at 25 d of culture, and the regeneration rate increased and reached 97.3% at 45 d of culture. Therefore, the regenerative pathway for F. nilgerrensis leaf discs was indirect organogenesis and embryogenic callus induction is an effective way to build an efficient regeneration system; here, CH plays a key role. The formation of embryogenic callus was the key to the substantial increase in the regeneration rate of F. nilgerrensis leaf discs, which guaranteed the regeneration of putative transgenic shoots.
Efficient shoot regeneration and rigorous antibiotic screening will considerably reduce the possibility of chimeric shoots in the genetic transformation of strawberries and facilitate the recovery of transgenic plants[20,44]. In our study, based on efficient shoot regeneration (97.3%), the transformation percentage for F. nilgerrensis leaf discs reached 8.67%, and PCR-positive transgenic lines were obtained in 16–20 weeks, which significantly improved over 20−24 weeks[15]. This may be due to the high-efficiency regeneration system based on embryonic callus development, which might boost the efficiency of selecting and proliferating single transformed cells, thereby reducing fewer chimeras and increasing the transformation efficiency[45]. Simultaneously, non-transgenic plants and false positive plants were eliminated by using different Kan concentrations; at Kan concentrations greater than 10 mg·L−1, non-transgenic leaf disc regeneration was severely hampered, which was in line with the findings of other studies[13]. However, a concentration of 20 mg·L−1 achieved efficient genetic transformation while avoiding the risk of chimerism or false positive plants in our experiment, which was lower than the value (25%−50%) reported in 'Chandler' by Husaini[20]. These differences are most likely due to the cultivars with varying levels of antibiotic tolerance.
-
In this study, the high-efficiency regeneration system for F. nilgerrensis leaf discs with a 97.3% regeneration rate was established on the MS medium with 2.0 mg·L−1 TDZ, 0.2 mg·L−1 IBA, and 0.2 or 0.5 g·L−1 CH in the dark culture for 14 to 21 d after optimizing several regeneration factors, which guaranteed the regeneration of putative transgenic shoots. By paraffin section observation of the callus from the leaf disc regeneration, the light yellow callus was determined to be embryogenic and the regenerative pathway was identified as indirect organogenesis. Furthermore, an average 8.67% transformation percentage was achieved by screening Kan concentration and referring to transformation procedures described by predecessors and PCR-positive transformants were obtained within 4−5 months. These findings will speed up studies on the gene functions and trait improvement of this species.
-
The plants of F. nilgerrensis (accession code SN11-6) were collected from Yiliang County (25°4'16" N, 103°13'16" E), Kunming City, Yunnan Province (China), in 2019 and grown in the open field of Shenyang Agricultural University (41°49'48" N, 123°34'12" E). Then 2−3 cm long runner-tips of F. nilgerrensis were collected, disinfected with 75% (v/v) alcohol for 10 s and 0.1% (v/v) mercuric chloride for 15 min in turn, and then rinsed three times with sterile and distilled water. The 0.5−1.0 mm apex meristems of the runners were then inoculated on MS media to grow into plantlets. The tube plantlets used for leaf regeneration were allowed to proliferate on the subculture medium MS + 0.5 mg·L−1 6-BA + 0.2 mg·L−1 GA3 (gibberellic acid). These plants on the subculture media were maintained in a growth chamber at 25 ± 2 °C under a 16 h photoperiod with 250 mol·m−2·s−1 illumination intensity and sub-cultured every four weeks.
Optimization of leaf regeneration conditions in F. nilgerrensis
-
Regeneration trials were performed using young expanded leaves detached from the tube plantlets. The leaf was cut transversely along the mid-vein to obtain the leaf discs of 0.8−1.2 cm as explants; these leaf discs were placed with the downward abaxial surface in contact with the regeneration media. A total of 34 assays with various combinations of treatments were designed, including different plant growth regulators (PGRs) (Table 1), dark culture durations (0, 7, 14, 21 and 28 d) and CH concentrations (0.0, 0.2, 0.5, 0.7 and 1.0 g·L−1), to optimize the regeneration system.
Thirty explants were maintained as three replicates for each treatment. After 45 d of culture, the percentages of callus induction and shoot regeneration were calculated as follows, and the number of shoots per leaf explant was measured. Callus induction rate (%) = (No. of leaf discs induced callus/ No. of inoculated leaf discs) × 100; Shoot regeneration rate (%) = (No. of explants regenerated shoots/ No. of cultured explants) × 100.
Observation of callus paraffin section
-
Paraffin section of the callus cultured for 25 d was used for further observation. The callus was fixed in formaldehyde–acetic acid–ethanol (FAA) solution for 2−4 d, dehydrated, embedded and stained[31]. The stained sections were observed, and images were captured using an Olympus microscope (SZX10, Olympus Corporation, Japan).
Agrobacterium strain and plasmid used for genetic transformation
-
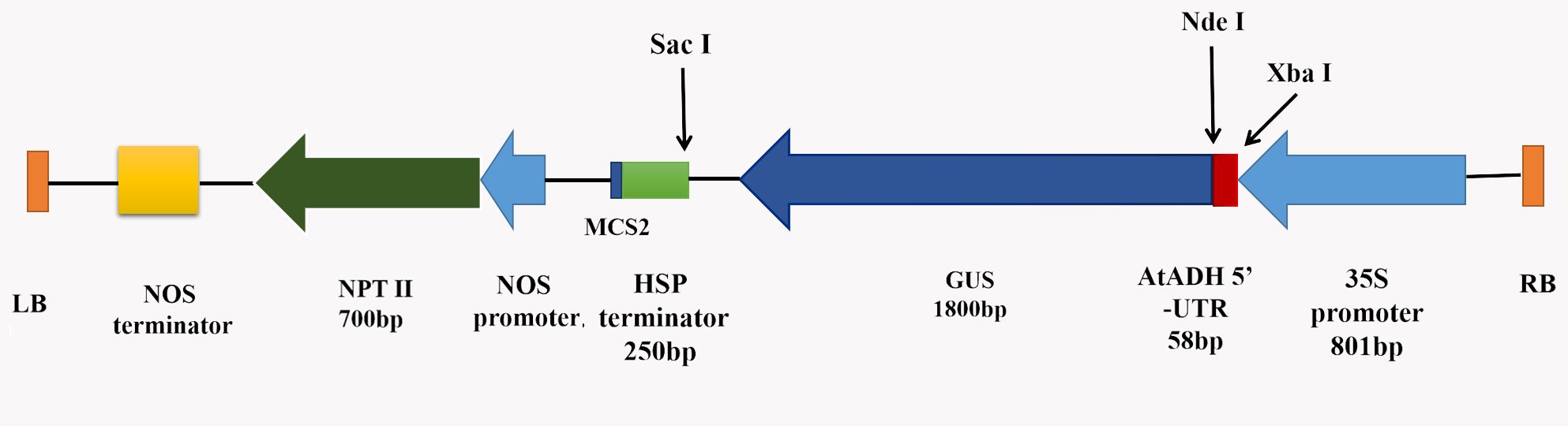
A binary vector pRI201-AN-GUS (TaKaRa, Dalian, China) (Supplemental Fig. S1), carrying the kanamycin (Kan) resistance gene NPTII (Neomycin phosphotransferase II) as a selection marker and the CaMV 35S promoter-driven GUS reporter gene, was used for genetic transformation. The Agrobacterium tumefaciens strain GV3101 was used for this process [46].
Optimization of kanamycin concentrations and Agrobacterium-mediated transformation of F. nilgerrensis leaf discs
-
The method of Agrobacterium-mediated genetic transformation for F. nilgerrensis leaf discs was referred to the previously described procedure with a slight modification[20].
The GV3101 cells transformed with the pRI201-AN-GUS plasmid were cultured in a YEP solid medium containing 50 mg·L−1 Kan and 20 mg·L−1 rifampicin (Rif) overnight at 28 °C on a rotary shaker (180 rpm·min−1). A single colony was confirmed by PCR using F: AGTCACGACGTTGTA and R: CAGGAAACAGCTATGAC primers, chosen, injected into 2 mL YEP liquid medium with the same antibiotics and cultured for 12−16 h at 28−30 °C on a rotary shaker (180 rpm·min−1). Then, 2 mL of the bacterial culture was added to 50 mL YEP liquid medium and cultured for 6−8 h to an OD600 value of 0.5−0.6 under the same antibiotics, temperature and speed. Then, the bacterial culture was centrifuged at 5000 r·min−1 for 5 min, and the obtained bacterial pellet was diluted in MS liquid medium (Table 5) to an OD600 value of 0.5. Finally, the bacterial suspension was mixed with 100 μmol·L−1 acetosyringone (AS) for explant infection. The whole processes of genetic transformation were co-culture, delayed-selection culture, selection and regeneration culture, shoot elongation culture and rooting culture, respectively.
Table 5. Composition of media used for genetic transformation of F. nilgerrensis leaf discs.
Procedure Media composition Duration Agrobacterium infection MS liquid medium, 100 μmol·L−1 AS, 5 g·L−1 glucose and 15 g·L−1 sucrose 10 min Co-culture MS, 2.0 mg·L−1 TDZ, 0.2 mg·L−1 IBA, 0.5 g·L−1 CH, 7 g·L−1 agar, 5 g·L−1 glucose and 30 g·L−1 sucrose 3 d Delayed-selection culture MS, 2 mg·L−1 TDZ, 0.2 mg·L−1 IBA, 0.5 g·L−1 CH, 7 g·L−1 agar, 30 g·L−1 sucrose, 250 mg·L−1 Tim and 250 mg·L−1 Cef 4 d Selection and regeneration culture MS, 2.0 mg·L−1 TDZ, 0.2 mg·L−1 IBA, 0.5 g·L−1 CH, 7 g·L−1 agar, 30 g·L−1 sucrose, 250 mg·L−1 Tim, 250 mg·L−1 Cef and 10/20 mg·L−1 Kan 60 d Shoot elongation culture MS, 2.0 mg·L−1 TDZ, 0.2 mg·L−1 IBA, 0.5 g·L−1 CH, 7 g·L−1 agar, and 30 g·L−1 sucrose, 250 mg·L−1 Cef and 10 mg·L−1 Kan 60−90 d Rooting culture 1/2 MS, 0.1 mg·L−1 IBA, 15 g·L−1 sucrose and 5 g·L−1 agar 30 d For co-culture, green leaves from 20-day-old plantlets were cut into leaf discs. The explants immersed in bacterial solution directly were gently shaken every 2 min for 10 min, dried on sterile filter paper, and then cultured in Petri dishes (100 mm × 25 mm) containing a co-culture media with the abaxial side up (Table 5). The plate was sealed with parafilm and incubated at 25 ± 2 °C in the dark for 3 d.
After 3 d of co-culture, explants were washed in a liquid co-culture medium supplemented with 250 mg·L−1 cefotaxime (Cef) and 250 mg·L−1 timentin (Tim), rinsed with vigorous shaking to remove the Agrobacterium cells, and then blotted dry on sterile filter paper. After washing, the explants were transferred to a delayed-selection media for 4 d (Table 5) and cultured under the same conditions as mentioned earlier.
The different concentrations of Kan (0, 5, 10, and 15 mg·L−1) were screened to determine the threshold concentration for eliminating non-transformed explants. Three replicates with 60 non-transformed explants each were used. The regeneration rate was calculated at 45 d of culture. After 4 d of delayed-selection culture, infected explants were transferred to a fresh selection and regeneration media with 10 mg·L−1 Kan for the selection of transformed cells and to inhibit further Agrobacterium growth for 14 d (Table 5). Further, to evaluate the effects on transformation efficiency, treatments with different Kan concentrations were used in this assay. After 14 d selective regeneration culture, the explants were subcultured on the selective media with different concentrations of Kan (10, 20, 30, 40 and 50 mg·L−1) for eliminating false positive transformed shoots for more than 4 weeks and the regeneration rate of the resistant shoot was calculated at 65 d post-infection. Sixty infected explants were cultured with three replicates per treatment.
The regeneration shoots were then subcultured on a selective elongation media with 10 mg·L−1 Kan and 250 mg·L−1 Cef for 8−12 weeks, and the transformation percentage was determined through PCR analysis at 130 d post-infection. After elongation culture, 4−5 cm long shoots were transferred to root induction medium for rooting (Table 5), and then transferred into 72-hole plug trays, followed by 10 cm × 10 cm pots, and finally planted in the open field. All the cultures were kept in the culture room at 25 ± 2 °C. Regeneration rate of resistant shoots (%) = No. of explants regenerated / No. of infected explants × 100; Transformation percentage (%) = No. of PCR-positive transgenic lines / No. of infected explants × 100.
To test the system's stability, 600 leaf discs with three replicates were used for genetic transformation. The regeneration rate of resistant shoots at 65 d post-infection and transformation percentage at 130 d post-infection was calculated.
Confirmation of transgenic lines
-
Polymerase chain reaction (PCR) analysis was performed using putative-resistant transgenic lines to detect the GUS marker gene and confirm the positive transgenic lines. One plantlet was randomly selected from each line for the experiment. Genomic DNA was extracted from leaves using a DNA extraction kit (Aidlab, Beijing, China). DNA samples from non-transgenic wild-type plants and ddH2O were used as negative controls, while the overexpression binary vector pRI201 plasmid acted as a positive control. The PCR to amplify the GUS gene (1800 bp) was performed in a 20 μL reaction mixture consisting of 1 μL of each primer (GUS-F: 5'-ATGTTACGTCCTGTAGAAAC-3' and GUS-R: 5'-TCATTGTTTGCCTCCC TGCT-3'), 10 μL 2×Taq Master Mix (Vazyme, Nanjing, China), 7 μL distilled sterile water, and 1 μL DNA (100 ng) on a PCR system with the conditions of denaturation (95 °C for 1 min), amplification with 28 cycles (95 °C for 15 s, 55 °C for 30 s and 72 °C for 30 s) and extension (72 °C for 7 min) in turn, following the method by Li et al.[13].
Histochemical GUS assay was performed using a GUS staining kit (Biosharp, Anhui, China) with PCR-positive transgenic lines to confirm the transgenic lines following the method by Li et al.[13]. Wild-type plants were used as control.
To further segregate Kan resistance PCR-positive transgenic lines and identify the transgene integration, the regeneration rates of leaf discs from the progeny of primary transgenic lines were assessed. Leaves of transgenic lines were randomly selected and cut into leaf discs to be inoculated on the regeneration medium with 10 mg·L−1 Kan for 2−3 weeks in the dark. Thirty leaf discs with three replicates were used. The leaves of the wild-type plants were used as the control. The regeneration rate was determined after 45 d of culture.
Statistical analysis
-
Data significant differences were calculated using the IBM SPSS Statistics 20 test and significant differences were set at P < 0.05.
This work was supported by Liaoning Provincial Science and Technology Project of 'Jiebangguashuai' (No. 2022JH1/10400016) and Shenyang Academician and Expert Workstation Project (2022-15).
-
The authors declare that they have no conflict of interest.
- Supplementary Fig. S1 Schematic of pRI201 vector.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Jiang S, Ji Y, Wang M, Xue L, Zhao J, et al. 2023. Establishing a high-efficiency in vitro regeneration system for Agrobacterium-mediated transformation in Fragaria nilgerrensis. Fruit Research 3:9 doi: 10.48130/FruRes-2023-0009 

Establishing a high-efficiency in vitro regeneration system for Agrobacterium-mediated transformation in Fragaria nilgerrensis
- Received: 27 November 2022
- Accepted: 01 March 2023
- Published online: 14 April 2023
Abstract: Fragaria nilgerrensis Schlechtendal ex J. Gay, a diploid wild strawberry, has many excellent characteristics such as fruit with white color and peach fragrance, and strong disease resistance. However, the lack of an efficient and stable regeneration and genetic transformation system for F. nilgerrensis has largely limited the functional studies of related genes governing excellent traits. In this study, a regeneration system for F. nilgerrensis leaf discs was developed by optimizing factors such as different hormone combinations, dark culture times and casein hydrolysate (CH) concentrations, with an average regeneration rate of 97.3% at 45 d of culture. By paraffin section observation of callus with different colors induced from the leaf disc regeneration, light yellow callus was determined to be embryogenic and the regenerative pathway was identified as indirect organogenesis. Based on this, an average transformation percentage of 8.67% was achieved by screening kanamycin concentration and referring to transformation procedures described by predecessors. PCR-positive transformants were obtained within 4−5 months by confirmation of PCR and histochemical GUS, and transgene integration was identified by transformants regeneration. The establishment of an efficient regeneration system provided a feasible platform for genetic transformation, which provided the foundation for further gene functional studies in F. nilgerrensis.
-
Key words:
- Regeneration /
- Genetic transformation /
- Fragaria nilgerrensis /
- Strawberry
{kind=link}