








-
As sequestered organisms, plants can enhance their environmental adaptation only when subjected to abiotic stresses through relevant physiological responses[1,2]. Environmental stresses to which plants are susceptible include temperature, light, UV, and soil pollution, etc[3]. Many fantastic phenotypes appeared when plants and environmental factors were associated. When winter arrives, plants resist the cold weather by slowing or stopping their growth[4,5]. Plants that are widespread in arid areas, such as Cactaceae, exhibit spiny leaves that reduce water evaporation, and roots are widely distributed underground, which increases water absorption[6,7]. Plants distributed at high altitudes sometimes exhibit developed glandular hairs, narrow leaf surfaces, and bright colors with higher anthocyanin content[8,9]. It is noteworthy that, on a more subtle level, the relationship between plant secondary metabolites and environmental factors has received increasing attention in recent years.
The model of 'environmental factors - phytohormones - transcription factors - secondary metabolites' as a regulatory network in medicinal plants has been established[10]. Plant secondary metabolites are not only valuable sustainable resources for human beings, but also play a key role in plants and the external environment[11]. Plant secondary metabolites, such as flavonoids, terpenoids, alkaloids and other natural products, have been reported to closely correlate with environmental factors[12]. Unfavorable environmental stresses and climatic factors, including drought, temperature extremes (freezing and high temperatures), light levels, nutrient deficiencies, and soil contamination with high concentrations of ions (metals and salts) are the main stressors affecting plant physiology during plant growth[13]. Metal elements are essential components in the soil environment and are necessary for the growth and development of plants. Zn, Cu, Fe and other elements are essential nutrients for plants, while the presence of Cd, Pb, and Hg in the soil will not only have a toxic effect on plants, but also endanger the food safety for humans[14]. Studies related to plant physiological effects of Fe date back to the mid-19th century, and Fe is involved in several significant physiological reactions[15,16]. Iron (Fe) is a mineral nutrient essential for plant growth and development and for various biochemical processes such as photosynthesis, respiration, and chlorophyll biosynthesis[17]. When deficient in iron, plant leaves usually develop interveinal chlorosis. However, iron is redox-active and therefore prone to the production of reactive oxygen species when it is present in excess in its free state[18]. Consequently, to maintain stable growth and development, plants are needed to ensure the distribution and efficient utilization of Fe in their tissues and organs[16].
Conyza blinii (C. blinii) is a traditional medicinal herb plant distributed in southwest China. The primary biological environmental conditions are dry-hot valleys, which suffer from drought and water loss, leading to high metal content in the soil[19]. Triterpenoids, mainly including blinin and Olas, are considered to be medicinal ingredients within C. blinii. In our previous study, we found that exogenous Fe2+ could increase the medical quality of C. blinii, as evidenced by enhanced photosynthetic efficiency, increased glandular trichome density, and improved blinin and total triterpenoid saponin contents[20,21]. In this experiment, on the basis of studying the correlation between Olas and Fe2+ in C. blinii, we further explored whether there is intratissue-targeted transport produced by Fe2+ stimulation after root splitting.
-
C. blinii were collected in 1/2-strength Hoagland's solution for further cultivation when they grew to 2 months old. The temperature was 26 ± 2 °C, and the photoperiod was 16 h light/8 h dark. The relative humidity was 50%−70%. The real leaves, tender stems and main roots were selected for testing.
Fe2+ treatment and split-root experiments
-
Referring to a previous research method, the concentration of Fe2+ was 200 μM[21]. The root system of C. blinii was equally divided into two parts, half of which was placed in 200 μM Fe2+ solution and the other in Hoagland nutrient solution, with no exchange of substances between the two parts. Group (0/0) which indicated no Fe2+ contact with either side of the root system. Group (Fe/Fe) which indicated Fe2+ contact with either side of the root system. In the split-root group (Fe/0), (Fe/0-Fe) indicates roots in direct contact with Fe2+ and (Fe/0-0) indicates roots not in contact with Fe2+. The split-root system is shown in Fig. 1. The experimental treatments lasted for 7 d, of which 3 d were replaced with new culture solution.
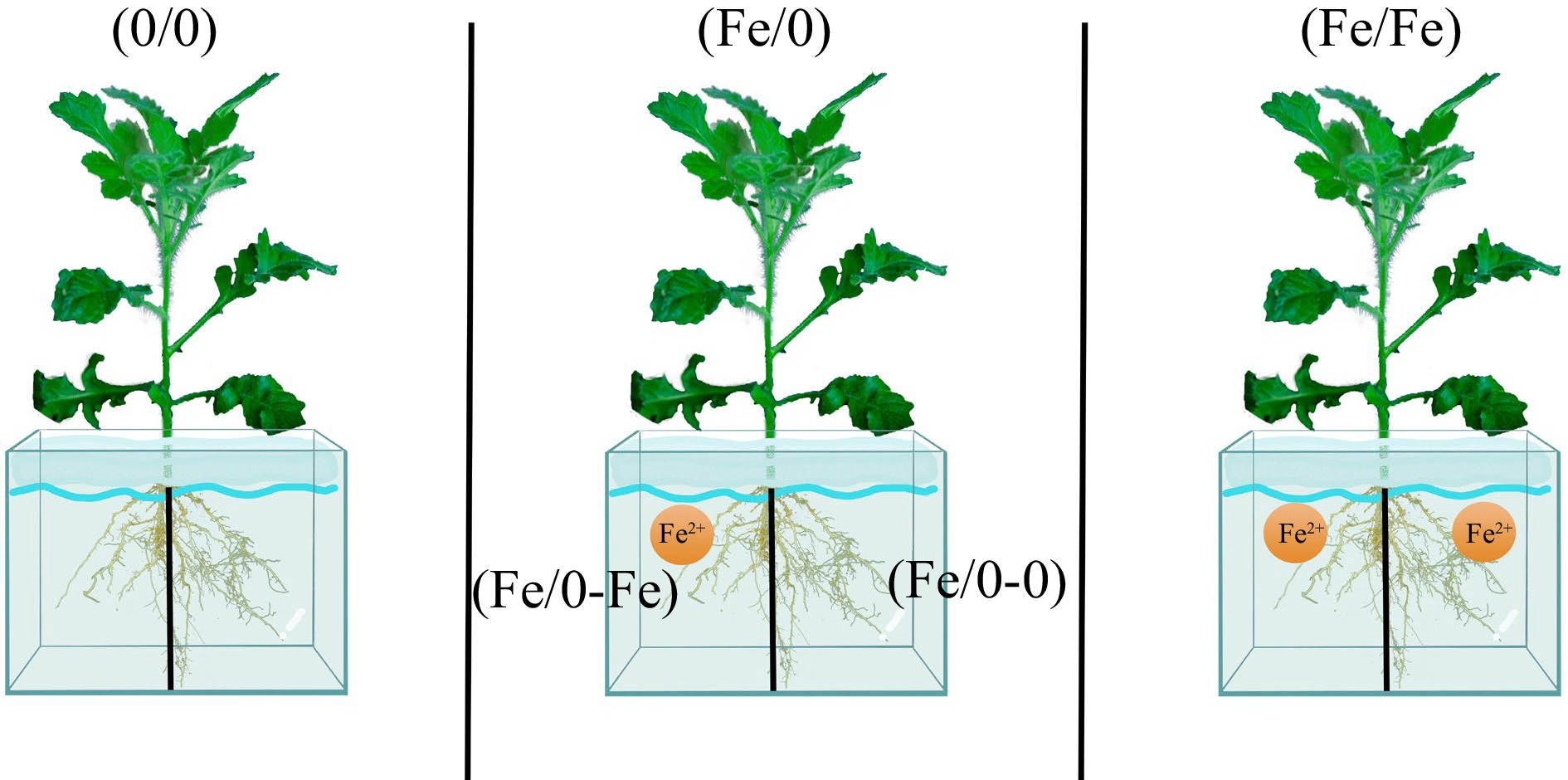
Figure 1.
Demonstration of root split-root experiments.
Determination of blinin and Olas
-
Leaves (0.05 g) were ground into powder using liquid nitrogen, and 500 μL of methanol was added for overnight maceration at 37 °C. Referring to previous research, HPLC was used to determine the blinin content at a detection wavelength of 210 nm[20]. Leaves (0.1 g) were rapidly ground in liquid nitrogen until powdered. The detection method of Olas was as described in our previous study[22]. Vanillin-perchloric acid method is used to detect the content of Olas. Determine the absorbance of the solution at 544 nm.
Determination of Fe content in plant tissues
-
Refer to Nanjing Jiancheng Tissue Iron Assay Kit (A039-2-1) for determination of plant iron content. The detection method of Fe content in plant tissues was as described in our previous studies[20,21].
RNA extraction and RT-qPCR analysis
-
Plant RNA was obtained using the EASYspin Plant RNA Rapid Extraction Kit (Beijing Adler Biotechnology Co., Ltd., RN2802, China) and reverse transcription was established by the FastKing One-Step RT-PCR Kit (Beijing Tiangen Co., KR123, China). A ChamQ Universal SYBR qPCR Master Mix kit (Nanjing Vazyme Q711-02, China) was used for the RT-qPCR experiments. Reaction systems, processes and data analysis methods were carried out as mentioned in our previous study[23].
Statistical analysis
-
We carried out analysis of variance (ANOVA) for all data, differences among treatments were analyzed by one-way ANOVA and differences were considered significant when p < 0.05, indicated by different letters. The experimental data were plotted with GraphPad Prism 9.0 software (
www.graphpad.com/features ). -
To initially investigate the effect of Fe on the metabolism of Olas, observations on samples 0−7 d after Fe treatment indicated that split-root leads to a decrease in iron absorption efficiency. Split-root reduced the 'poison' phenotype of leaves caused by iron absorption (Fig. 2a), effectively reducing the iron content in leaves by 30%–32% at the end of the treatment (Fig. 2b). There was a significant difference in Fe content in the roots of the (Fe/0) group. The Fe content in the (Fe/0-Fe) group reached 185 mg·g−1 at 7 d, while it was only 22 mg·g−1 in the (Fe/0-0) group.
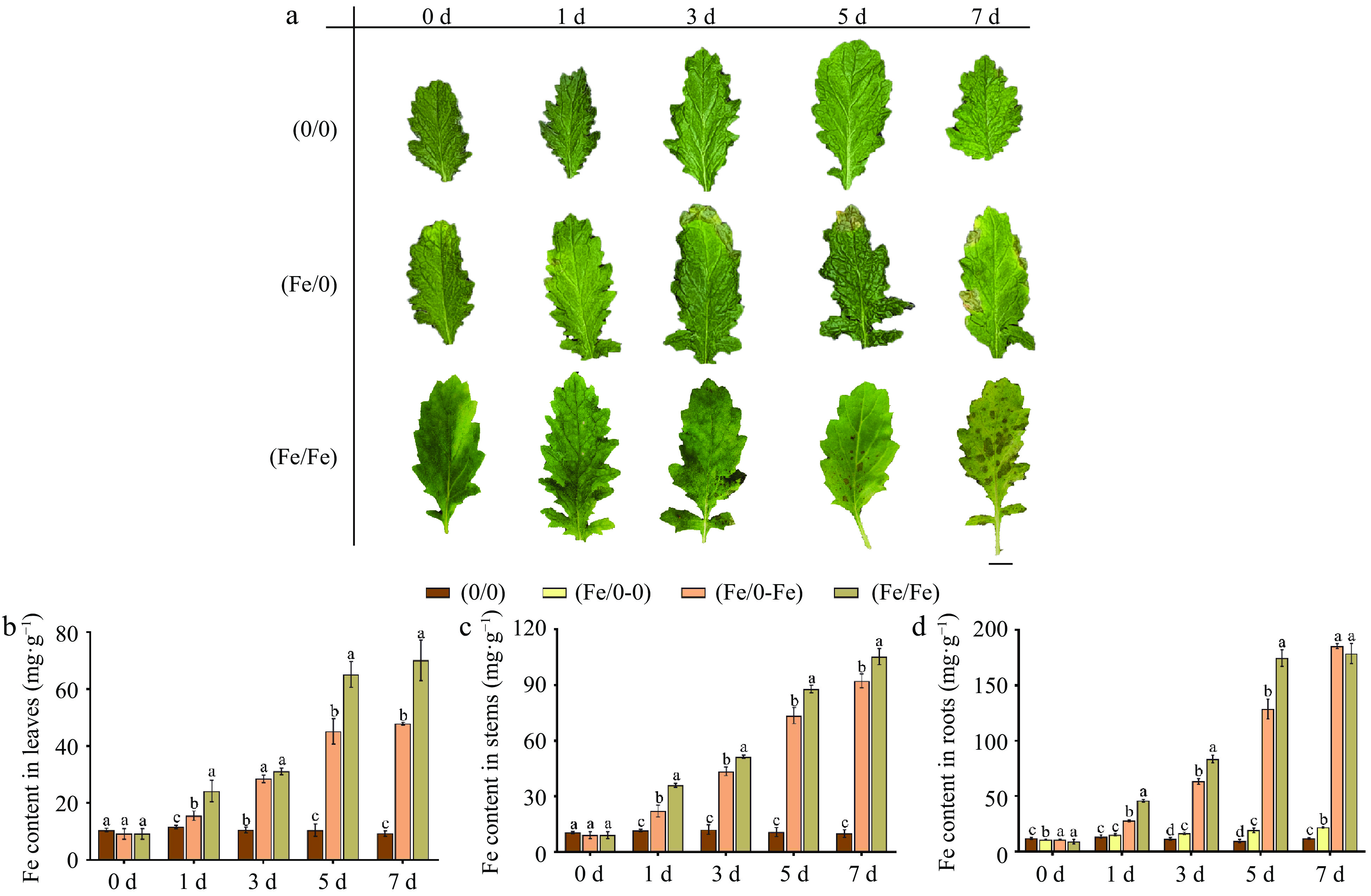
Figure 2.
(a) Leaf phenotypes in split-root system under Fe2+ treatment of C. blinii. Bar = 1 cm. Fe content in (b) leaves, (c) stems and (d) roots in split-root system under Fe2+ treatment. The concentration of Fe2+ treatment was 200 μM. All experiments were performed using at least three biological replicates and error bars indicate standard deviations (± SD). Different letters indicate significant differences at the p < 0.05 level when comparing different experimental groups.
Oleanolic acid content in leaves decreased when roots were exposed to Fe2+
-
For further investigation, oleanolic acid content in different tissues with split-root was examined, which was used to indicate the interaction between Fe2+ and the MVA pathway. The oleanolic acid content in leaves showed a significant decrease when roots were exposed to Fe2+, decreasing from 0.89 mg·g−1 to approximately 0.40 mg·g−1 (Fig. 3a). In contrast, oleanolic acid content in roots presented a fluctuating increase, reaching a maximum value of 0.78 mg·g−1 at 4 d (Fig. 3c). It is notable that in roots, the variation in oleanolic acid content of the (Fe/0) group appeared to be dichotomous, while the (Fe/0-0) group varied similarly to the (0/0) group, as well as the (Fe/0-Fe) and (Fe/Fe) groups.
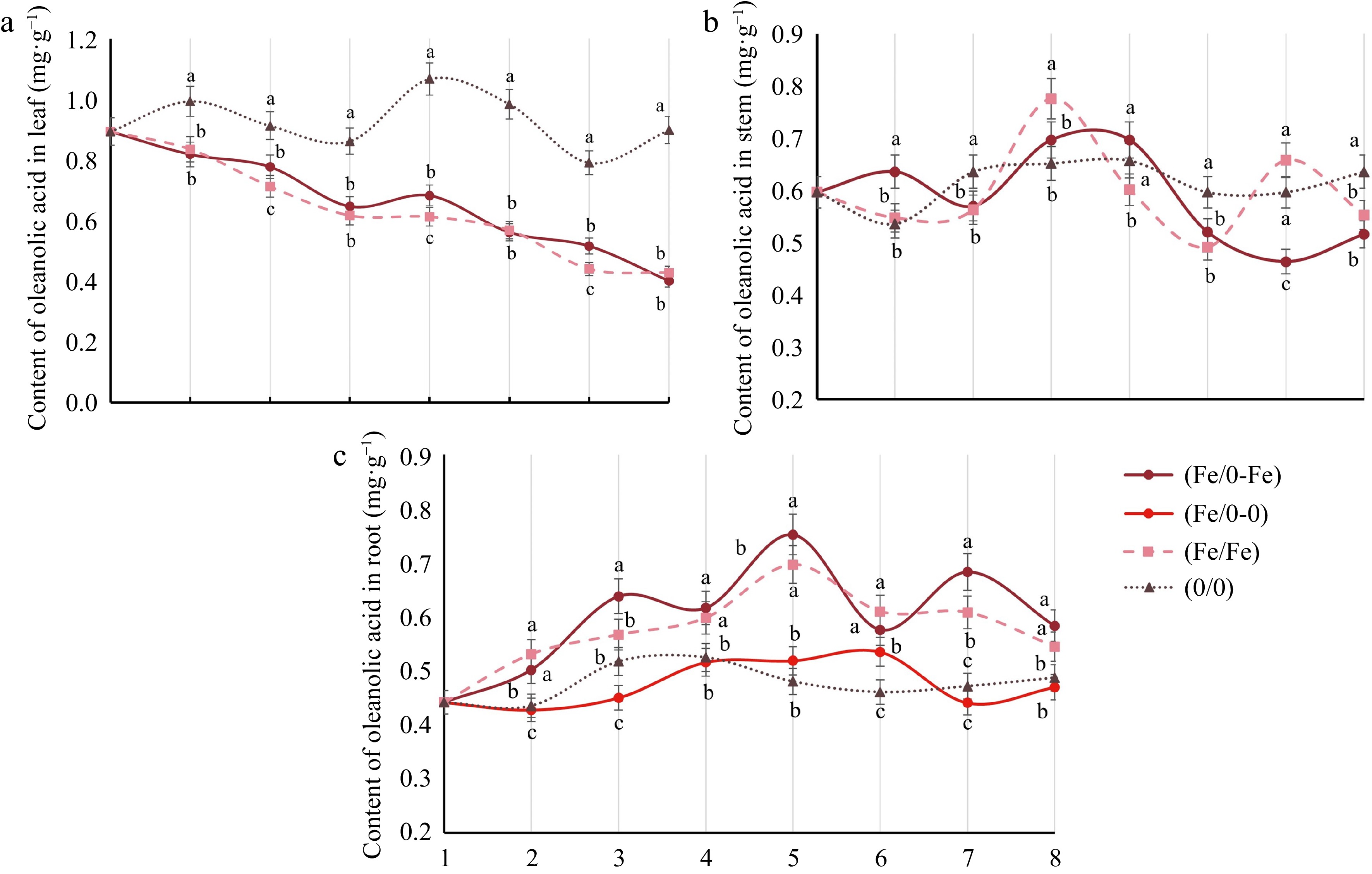
Figure 3.
Analysis of oleanolic acid in various tissues of C. blinii. Oleanolic acid in (a) leaves, (b) stems and (c) roots under Fe2+ treatment with split-root system. All experiments were performed using at least three biological replicates and error bars indicate standard deviations (± SD). Different letters indicate significant differences at the p < 0.05 level when comparing different experimental groups.
Fe2+ stimulates the expression of key enzymes genes of the MVA pathway
-
The stimulation of Fe2+ to the leaf MVA pathway was not affected by split-root, either in the (Fe/Fe) or (Fe/0) groups (Fig. 4a). The most significantly stimulated was CbHMGR, which showed a 5.59-fold increase in relative expression at 5 d in (Fe/Fe). There was a 3.49-fold increase in the relative expression of CbHMGR at 3 d in (Fe/0). The stimulation of MVA metabolism was stronger for (Fe/Fe) than (Fe/0) in leaves. However, in root tissues, the difference in the distribution of Fe2+ has brought a variation in the relative expression of MVA genes. The maximum relative expression values of CbHMGR were 7.13 and 2.49 in the (Fe/0-Fe) and (Fe/0-0) groups, respectively, while the maximum relative expression of the (Fe/Fe) group was 5.86 (Fig. 4c). The activity of MVA metabolism in stems also fluctuated due to the existence of Fe2+ (Fig. 4b). These results suggested that Fe2+ is a direct stimulating signal for the MVA metabolic pathway.
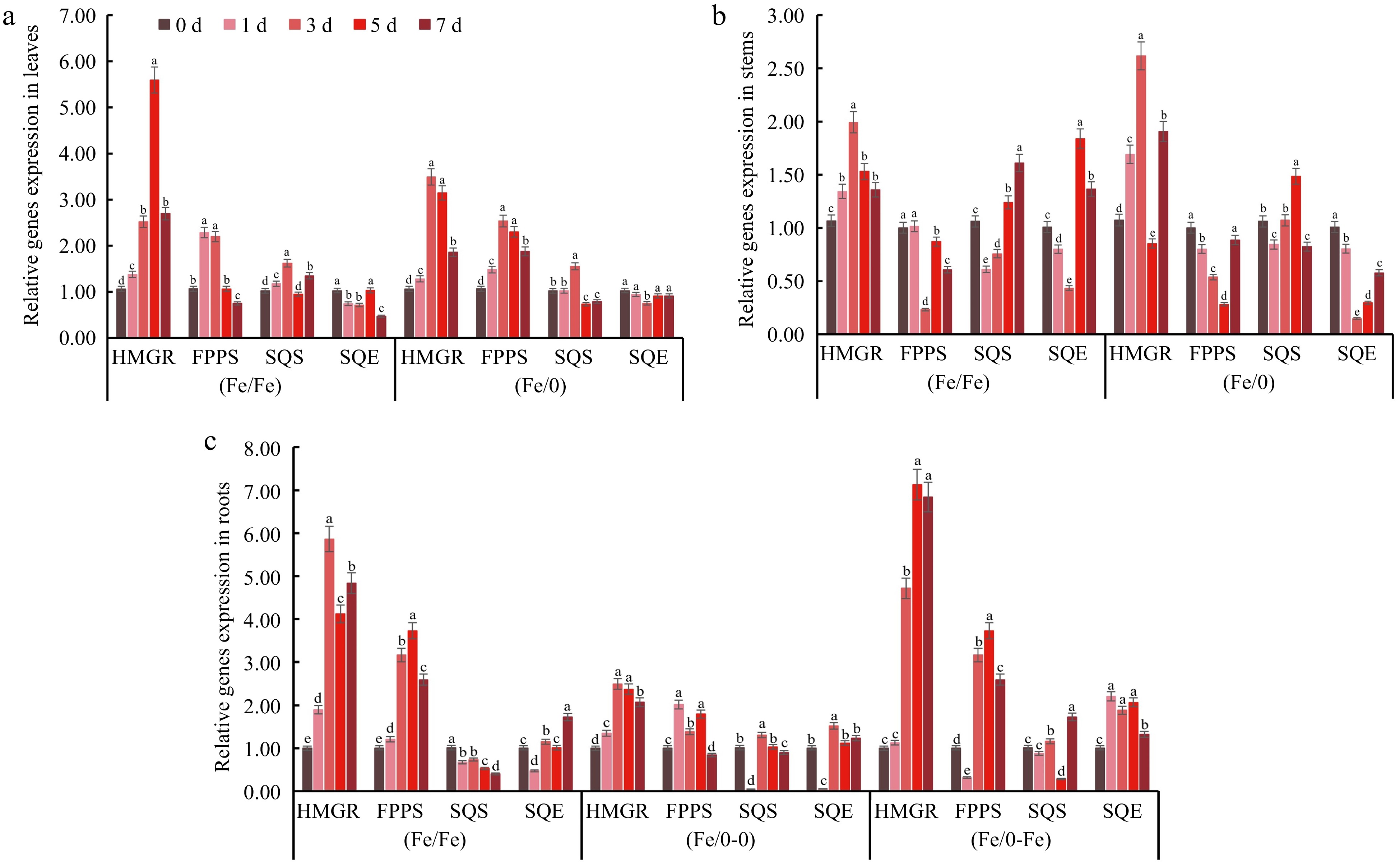
Figure 4.
Relative expression of key MVA pathway genes in various tissues. The genes relative expression in (a) leaves, (b) stems and (c) roots under Fe2+ treatment with split-root system. All experiments were performed using at least three biological replicates and error bars indicate standard deviations (± SD). Different letters indicate significant differences at the p < 0.05 level when comparing different experimental groups. The 2−ΔΔCᴛ method was used to determine the relative expression and the genes relative expression of (0/0) group were set to '1'.
Fe2+ stimulates blinin specifical accumulating in leaves
-
In comparison to oleanolic acid, the blinin content among tissues showed significant differences under Fe2+ treatment with split-root system. The blinin content increased from 0.24 to 0.55 mg·g−1 and 0.61 mg·g−1 in the (Fe/0) and (Fe/Fe) groups, respectively, whereas the blinin content in (0/0) group was maintained at approximately 0.18 mg·g−1 (Fig. 5). Nevertheless, blinin was almost undetectable in stems and roots (Table 1). From this, it can be hypothesized that in comparison to the global distribution of oleanolic acid, blinin specifically accumulates in the leaves of C. blinii.
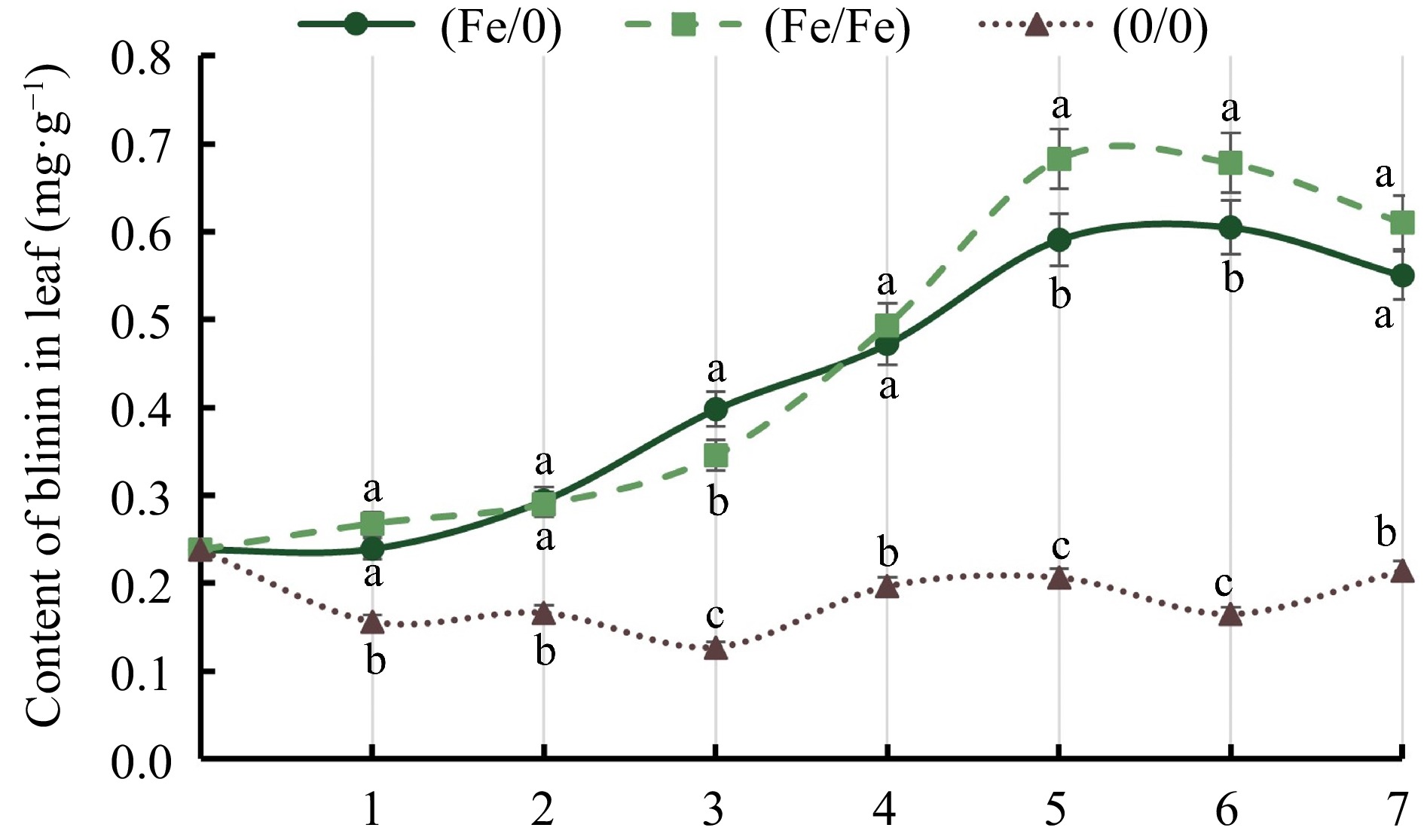
Figure 5.
Analysis of blinin in leaves of C. blinii. All experiments were performed using at least three biological replicates and error bars indicate standard deviations (± SD). Different letters indicate significant differences at the p < 0.05 level when comparing different experimental groups.
Table 1. Blinin content in stems and roots.
Sample 0 d 1 d 2 d 3 d 4 d 5 d 6 d 7 d Stems (0/0) 0.013872 0.011378 n.d. 0.02471 n.d. n.d. 0.012124 0.013872 Stems (Fe/Fe) 0.013872 n.d. n.d. 0.021454 n.d. 0.01274 n.d. 0.013872 Stems (Fe/0) 0.013872 0.010663 n.d. n.d. n.d. n.d. n.d. 0.013872 Roots (0/0) n.d. 0.011252 n.d. n.d. n.d. 0.01274 n.d. n.d. Roots (Fe/Fe) n.d. 0.009729 n.d. n.d. 0.0124 n.d. n.d. n.d. Roots (Fe/0-Fe) n.d. n.d. n.d. n.d. n.d. n.d. n.d. n.d. Roots (Fe/0-0) n.d. n.d. n.d. 0.010065 n.d. n.d. n.d. n.d. 'n.d.' represents that the blinin content in tissue samples didn't reach the minimum detection limit of HLPC. Unit: (mg·g−1). Fe2+ stimulates the expression of key enzyme genes of the MEP pathway
-
The promotion of Fe2+ on the MEP pathway has similarities to the MVA pathway. The stimulating effect on CbDXR was more obvious than CbDXS (Fig. 6a). The relative expression of CbDXR reached maximum values of 3.4 and 5.8 in (Fe/Fe) and (Fe/0) groups, respectively, in leaves. The highest relative expression of CbDXS was only 2 and 1.65. In addition, the relative expression of CbDXR reached maximum values of 3.25 and 3.34 in the (Fe/Fe) and (Fe/0-Fe) groups, respectively, in roots (Fig. 6c). The stimulating effect of Fe2+ on the expression of CbMCS and CbGGPPS remained essentially similar among the other groups. However, in the (Fe/0-Fe) group, the expression of CbDXS was downregulated by Fe2+ in split-root system. The activity of MEP metabolism pathway in stems also fluctuated due to the existence of Fe2+ (Fig. 6b). These results suggested that Fe2+ regulates the activity of the MEP pathway as well as MVA in the split-root system, which is consistent with our previous results[22]. The presence of Fe2+ encourages the activity of the MEP pathway enzyme-encoding, which is responsible for the increase in blinin content in leaves.
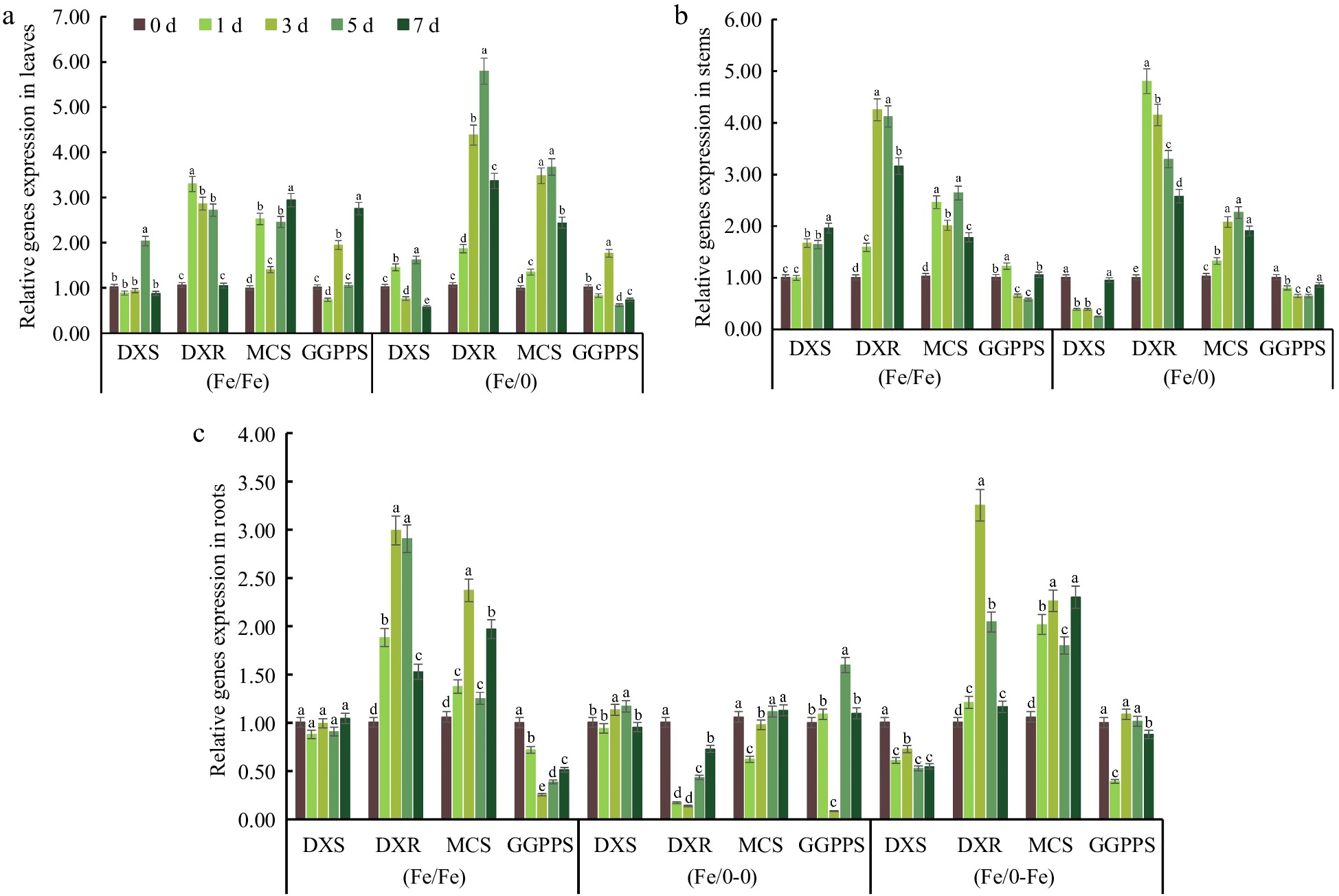
Figure 6.
Relative expression of key MEP pathway genes in various tissues. The gene relative expression in (a) leaves, (b) stems and (c) roots under Fe2+ treatment with split-root system. All experiments were performed using at least three biological replicates and error bars indicated standard deviations (± SD). Different letters indicate significant differences at the p < 0.05 level when comparing different experimental groups. The 2−ΔΔCᴛ method was used to determine the relative expression and the genes relative expression of (0/0) group were set to '1'.
PDR may be involved in the transport of Olas to roots under Fe2+ treatment
-
The presence of Fe2+ reduced the Olas content in leaves but increased it in roots, suggesting the possibility that Olas was transferred to roots. PDR genes have been reported to have the ability to transport terpenoids[24]. There was no significant fluctuation in the relative expression of CbPDR in the (0/0) group, but the peak values of CbPDR were 8.42 and 10.51 in the (Fe/Fe) and (Fe/0) groups in leaves. The maximum CbPDRs were 4.47 and 10.10 in the (Fe/Fe) and (Fe/0) groups in stems, respectively. Notably, the relative expression of CbPDR in the (Fe/0-0) group was significantly increased by Fe2+ in the end. Above all, it can be assumed that Olas was transported by PDR through leaves to roots, responding to the stimulation of Fe2+.
-
In this study, the metabolism and accumulation of blinin and Olas were explored by a split-root system under Fe2+. Split-root experiments have been widely used in research focused on understanding the complex regulatory mechanisms of legume-rhizobium symbiosis, root nitrogen rhizobium deposition and belowground nitrogen translocation, as well as the effects of different biotic/abiotic factors on such symbiotic interactions[25,26]. The split-root method has been employed to study the transport of phosphorus by wheat[27]. Luo et al.[28] treated one side of the cotton root system with PEG and found that the JA content of cotton leaves increased and was transported through the phloem to increase ABA synthesis in the root system that was not treated with PEG, up-regulated the expression of GhPIP, and promoted water uptake in the other side of root. Similarly, after treating one side of the roots with Fe2+, there was some increase of saponin content in the other side of the roots that were not exposed to Fe2+ (Fig. 3). The response of saponins to Fe may be tissue-specific, but it is possible that some saponins may 'get lost'.
MVA pathway and Olas may be the main defense against Fe2+
-
Fe2+ signaling modifies the original accumulation pattern of terpenoids in C. blinii. The accumulation of natural ingredients in medicinal plants is characterized by tissue variation. Artemisinin synthesis-related regulators are specifically expressed in glandular trichomes, while the accumulation of artemisinin in glandular trichomes is also higher than that in other tissues[29,30]. Plant secondary metabolites can be detected throughout the plant, but in some cases the site of biosynthesis is limited to a single organ and is transported to different storage sites via vascular tissues or symbiotic and non-symbiotic transport, depending on the polarity of the metabolites[31,32]. The yield and concentration of SMs produced by plants is determined by the balance between biosynthesis, storage and degradation, which depends on which secondary metabolites become the dominant phase[33]. The increased level of Olas in roots exposed to iron suggests that Olas seems more probable to be a defense against Fe2+ in C. blinii. Changes in Olas content precede those of blinin, and changes in the first step enzyme-encoding gene CbHMGR also precede that of CbDXS, revealing that the MVA pathway responds earlier than the MEP pathway to root Fe2+. MVA and MEP are two metabolic pathways located in the cytoplasm and chloroplast respectively[34]. MVA pathways may receive stimulatory effects from Fe2+ more quickly.
PDR transport was involved in the transportation of Olas
-
The transport of active ingredients is the basic protection for the maintenance of the vital activities of different tissues. In plants, terpenoids are usually transported from the cell in which they are synthesized to an adjacent cell or from the synthesized tissue to other tissues, which involves many transport proteins[35−37]. PDR transporter proteins are involved in plant defense through active transport of terpenoids through vesicular segregation and exocytosis to achieve intracellular and intercellular transport, AaPDR3 and NtPDR1 have been reported to be involved in terpenoid transportation[38,39]. In this experiment, CbPDR was upregulated in all tissues, suggesting that basic Olas transportation occurred (Fig. 7). As PDR expression was more active in groups (Fe/0) and (Fe/Fe), it is presumed that Olas was transported from leaves to the roots via the stem to adapt to Fe2+ signaling.
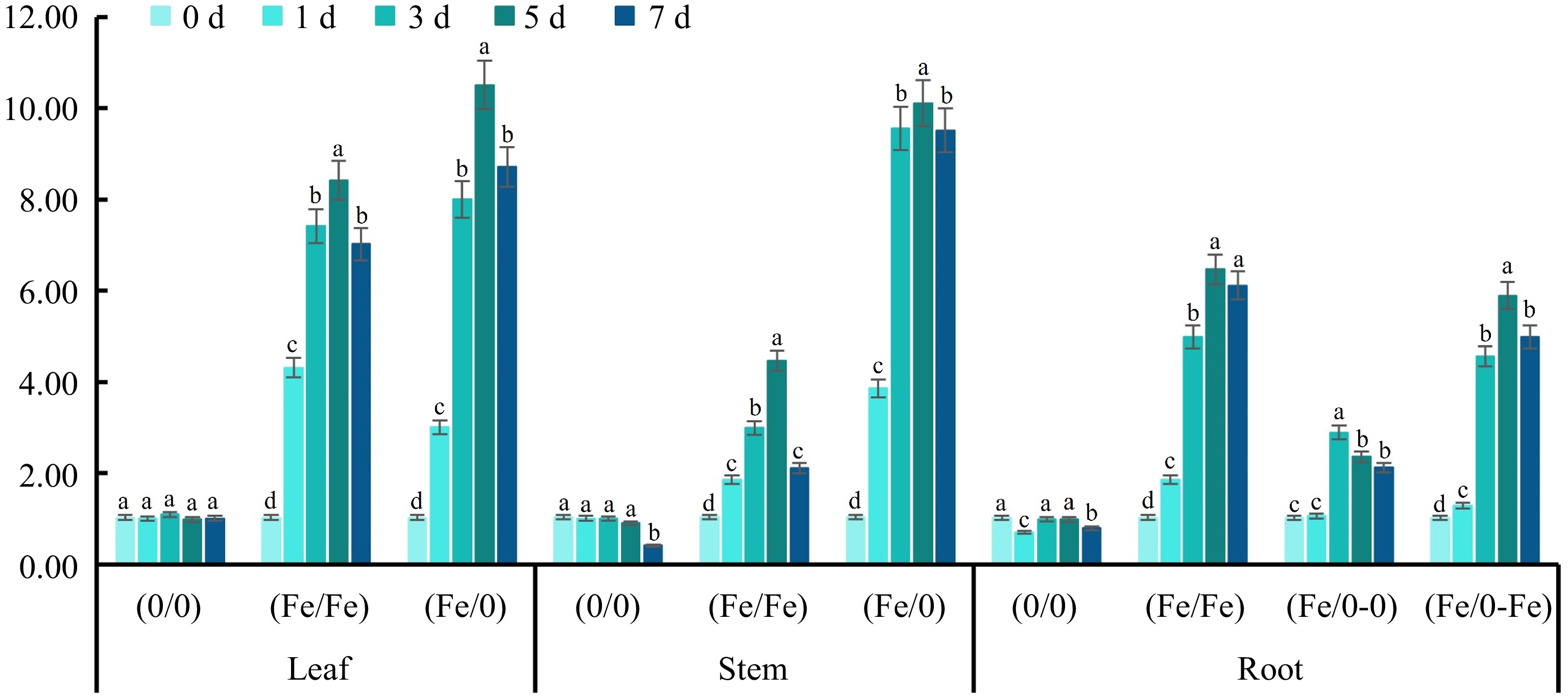
Figure 7.
Relative expression of CbPDR in various tissues. The relative expression of CbPDR in (a) leaves, (b) stems and (c) roots under Fe2+ treatment with split-root system. All experiments were performed using at least three biological replicates and error bars indicated standard deviations (± SD). Different letters indicate significant differences at the p < 0.05 level when comparing different experimental groups. The 2−ΔΔCᴛ method was used to determine the relative expression and the genes relative expression of (0/0) group were set to '1'.
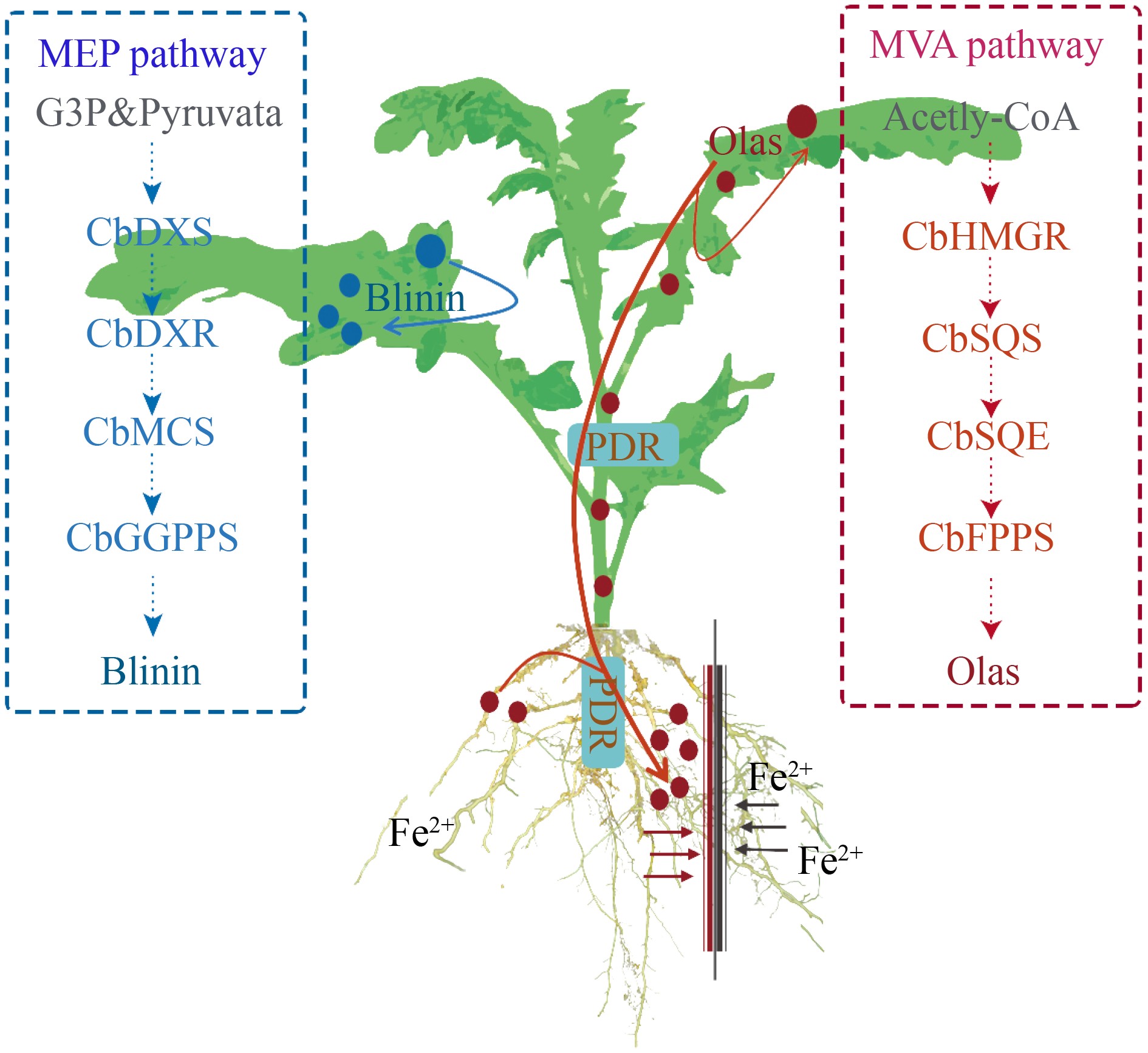
Overall, our studies revealed that root Fe2+ could modify the original cumulative pattern of blinin and Olas in C. blinii. On the one hand, blinin was accumulated specifically in leaves, which depended on the time of root Fe2+ treatment. On the other hand, Fe2+ induced Olas targeted translocation and accumulation in roots of C. blinii (Fig. 8).
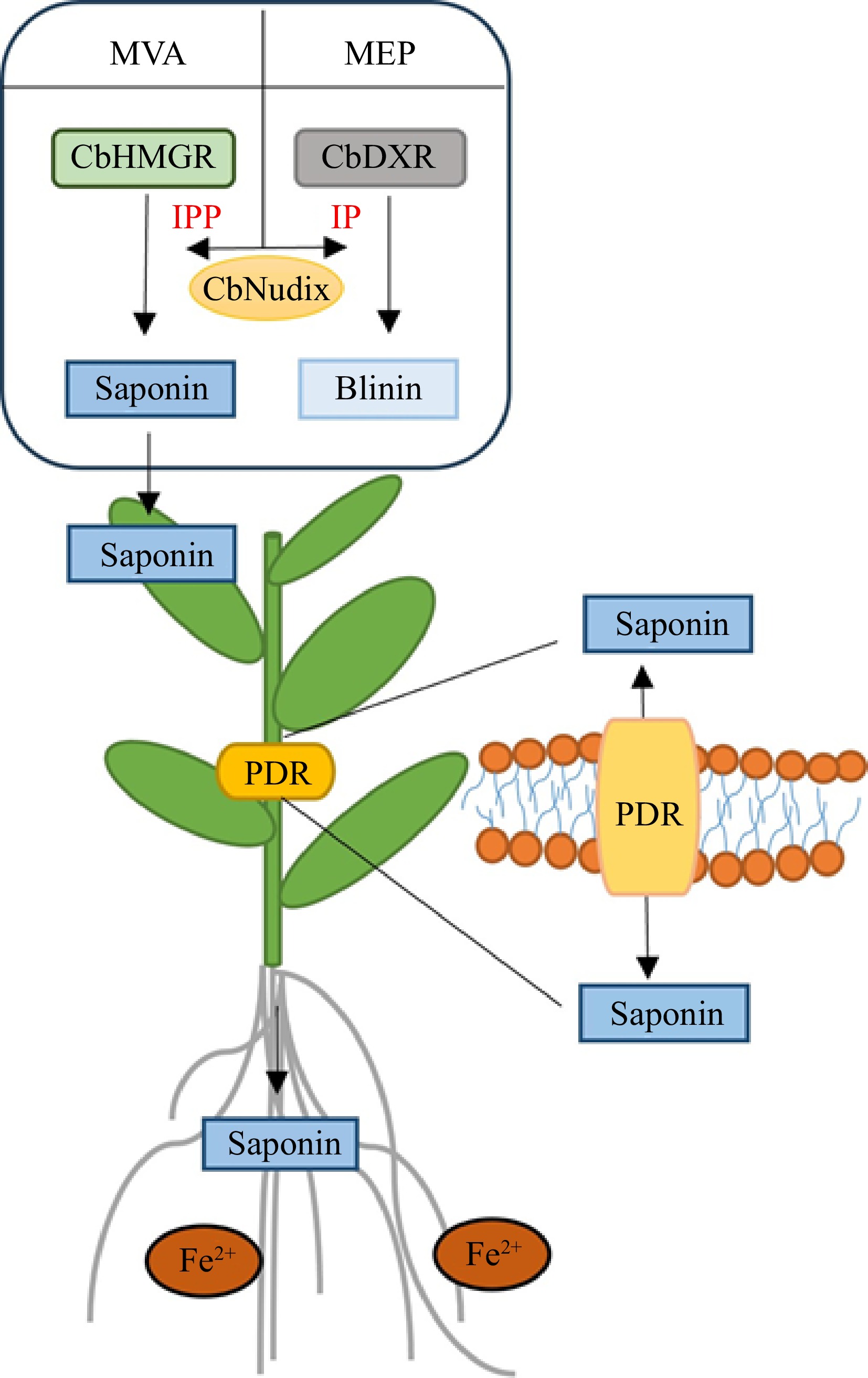
Figure 8.
A model for the transportation of triterpenoid saponins by PRD under Fe2+. Under Fe2+, the activity of the MEP metabolism pathway was enhanced within C. blinii. The solid line represents signal transduction.
-
The authors confirm contribution to the paper as follows: study conception and design: Chen H, Zheng T; research performed: Wang M, Yang M, Zhan J, Zheng T, Zhou M; data analysis: Liu M, Zheng T; project administration: Liu M, Zheng T; draft manuscript preparation: Yang M, Wang M. All authors read and approved the final manuscript.
-
The previous raw data of RNA-sequencing have been deposited in the Sequence Read Archive in National Genomics Data Center (
https://ngdc.cncb.ac.cn/ ) under accession number SRR10053375, SRR10053374 and SRR10053373. -
We thank all the colleagues in our laboratory for providing useful discussions and technical assistance. In particular, we would like to thank the Yongchuan District Center for Disease Control and Prevention for providing HPLC services. We are very grateful to the editor and reviewers for critically evaluating the manuscript and providing constructive comments for its improvement.
-
The authors declare that they have no conflict of interest.
-
Received 25 August 2023; Accepted 8 January 2024; Published online 26 January 2024
-
The activities of MEP/MVA pathway were stimulated in all tissues by the additional rhizospheric Fe2+.
Olas was transported from leaves to roots in response to Fe2+, which was demonstrated by the split-root system.
The cross-tissue transportation of Olas may be mediated by the PDR transporters.
-
# Authors contributed equally: Maojia Wang, Ming Yang
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Hainan University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Wang M, Yang M, Zhou M, Zhan J, Liu M, et al. 2024. Effect of rhizospheric Fe2+ on terpenoid biosynthesis and accumulation within Conyza blinii H. Lév. Tropical Plants 3: e003 doi: 10.48130/tp-0024-0003 

Effect of rhizospheric Fe2+ on terpenoid biosynthesis and accumulation within Conyza blinii H. Lév.
- Received: 25 August 2023
- Revised: 27 December 2023
- Accepted: 08 January 2024
- Published online: 26 January 2024
Abstract: Conyza blinii (C. blinii) is a herbaceous plant that is distributed in dry-hot valleys in southwest China. Metabolites of medicinal plants are commonly associated with environmental factors. Dissipation of soil water caused by heat increases soil metal levels. Oleanane-type triterpenoid saponins (Olas) are the main active medicinal ingredients in C. blinii. Here, we explore the effect of Fe2+ on terpenoid metabolism, especially the mevalonic acid (MVA) pathway. The results indicate that the activity of the MVA and methylerythritol phosphate (MEP) metabolic pathways are increased by Fe2+ and that the expression of key enzyme-encoding genes of metabolic pathways3-hydroxy-3-methylglutaryl coenzyme A reductase (CbHMGR), Farnesyl pyrophosphate synthase (CbFPPS) and 1-deoxy-D-xylulose-5-phosphate synthase (CbDXS) are also significantly upregulated. Moreover, the triterpenoid saponin content in the leaves gradually decreased, but in the roots, it increased. Furthermore, Olas were confirmed to be transported from leaves to the roots by the root-split system, which was accompanied by high-level expression of pleiotropic drug resistance (CbPDR). Above all, our experiments revealed that Olas of C. blinii were actively synthesized in the leaves and transported to the roots via the CbPDR in response to the stimulation of Fe2+.
-
Key words:
- Conyza blinii /
- Fe2+ /
- Terpenoid metabolism /
- Blinin /
- Saponin /
- Split-root /
- PDR