








-
The vesicle-associated membrane protein (VAMP)-associated protein (VAPs) family has been identified as a highly conserved group of proteins identified both in plants[1] and animals[2]. It was first identified in animals participating in the transportation of related substances by forming SNARE protein complexes[3]. Their plant homologues are named VAP27 because the first member identified had a molecular weight of 27 kDa[4]. The structure of VAPs usually contains an N-terminal major sperm domain (aa 1–129), a coiled-coil domain (aa 178–234), and a C-terminal transmembrane domain (aa 234–253)[4]. It is reported that the N-terminal sperm domain is crucial for the interaction between VAP27-1 and NET3C, and the fixation of VAP27-1 in the ER-PM contact site[5]. In Arabidopsis, 10 VAP homologs have been identified[1]. To date, numerous research on VAPs have been reported in the plant kingdom, and increasing proteins relevant to VAPs have also been discovered[5−8]. For example, NET3C and VAP27 form homo-dimers or homo-oligomers that function in mediating the communication of PM and ER via interacting with actin and microtubules for lipid transport, calcium influx, and other vital biological processes[5]. VAP27-1 and VAP27-3 have been revealed to interact with clathrin and play a central role in maintaining clathrin homeostatic dynamics at endocytic membranes and regulating endocytosis[8].
According to existing studies, VAP proteins family localize to the endoplasmic reticulum (ER) and ER/plasma membrane (PM) contact sites and are tightly linked to the cytoskeleton that plays a supporting role[9−12]. The endoplasmic reticulum (ER), as one of the important components of the intimal system, plays an indispensable role in protein synthesis, folding and quality control, protein secretion, lipid biosynthesis, and calcium storage[13,14]. The ER is also actively involved in endocytosis with mechanisms that have not been clearly defined[15]. The transportation and translocation of various proteins, steroids, lipids, and other synthesized molecules typically depend on ER-centered traditional vesicular trafficking pathways[16,17]. The close association between the ER and the PM, facilitated by proteins like VAPs, is essential for vesicle trafficking from the ER. Given the intimate connection between the VAP family and the plasma membrane, researchers propose VAP interactions with proteins involved in plant development and maintaining structural stability. This explains the requirement of VAPs for growth, cell division, and abiotic stress responses[18−22]. Recent research in Arabidopsis thaliana also supports this idea, revealing that SYT1, an ER-resident protein[23], plays a vital role in stabilizing the ER network and connection between VAP27-1-enriched ER and plasma membrane[24]. VAMP721/722 are components of the default secretory pathway and can transport substances required for cell growth, suggesting a potential role in plant autoimmune regulation.
There have been studies demonstrating that the plasma membrane participates in the secretion of immune protein for the activation of plant immune defense against pathogen invasion. For instance, the antimicrobial proteins secreted through vesicle trafficking was targeted and destroyed by the RxLR effector of Phytophthora brassicae by working together with host RABA-type GTPase, subsequently compromising the immune system[25]. There are also reports indicating that the VAP protein family could influence the development of various plants and defense networks. The immune mechanism of the VAP protein family in Arabidopsis thaliana and tomato has been confirmed[20,22].
Grapevine (Vitis vinifera L.) is distinguished as one economically valuable fruit, appreciated both for fresh consumption and the production of various processed items such as wine and grape juice. Environmental stresses can seriously affect grapevine growth and development in cropland. For instance, high humidity on prolonged rainy days during critical maturation stages can compromise the quality of grapevine, while drought conditions can drastically reduce fruit yields. Additionally, biotic stresses, such as downy mildew, powdery mildew, anthracnose, and others[26,27], pose threats by impeding normal leaf growth and causing yield losses. Given its substantial economic importance, grapevine cultivation is widespread across various countries. The identification of significant functional genes becomes of utmost interest.
At present, research on the VAP27 protein is limited, particularly in the context of grapevine. Therefore, in this study, we identified and analyzed the VAP27 protein family through bioinformatic analysis of genomic and transcriptomic data. The structure and function of the VAP27 gene were preliminarily analyzed, laying the foundation for further study of gene functionality.
-
The grape genome sequences of Vitis vinifera cv. 'Pinot Noir' (PN40024.v4) were downloaded from Ensemble Plants (
https://plants.ensembl.org/Vitis_vinifera/Info/Index ). Initial identification involved querying the grape genome database using the Arabidopsis VAPs protein sequences through BLAST. Next, an HMM file was constructed using the seed alignment file for the VAP domain (PF00635) obtained from the Pfam database, utilizing the HMMER3 software package. HMM searches were then performed against local protein databases of grape sequencing using HMMER3. To ensure accuracy, the physical localizations of all candidate Vitis vinifera VAP27s (VvVAP27s) on chromosomes were examined, and redundant sequences with identical chromosome locations were excluded. All obtained VAP27 protein sequences were subjected to Pfam analysis (http://pfam.xfam.org/ ) to verify the DBD domain. The presence of DBD domains and coiled-coil structures was confirmed using SMART (http://toolkit.tuebingen.mpg.de/marcoil ) and MARCOIL (http://toolkit.tuebingen.mpg.de/marcoil ). Sequences lacking the DBD domain or a coiled-coil structure were eliminated from further analysis.Phylogenetic, exon-intron structure, and motif analysis of VAP27 Genes
-
Genome sequences, CDS sequences, and protein sequences of the VAP27 family were downloaded for analysis. An unrooted phylogenetic tree was constructed for sequences from grapes, Arabidopsis thaliana, tomatoes, and rice using the Neighbor-Joining (NJ) method with the bootstrap test replicated 1,000 times. The software used for creating these phylogenetic trees was MEGA5. The exons and introns of grape VAP27 genes were determined based on alignments of transcribed sequences and corresponding genomic sequences, and the visualization of VAP27 gene structures was performed with the online Gene Structure Display Server 2.0. Conserved motifs and domains of grape VAP27 genes were identified using MEME 4.11.2 (
http://meme-suite.org/tools/meme ) and SMART (http://toolkit.tuebingen.mpg.de/marcoil ) software.Transcriptome analysis of VvVAP27 gene
-
Using published data[28], the expression patterns of VvVAP27 gene family at 54 stages of grape plant development were analyzed using the average expression values of three biological replicates. Clustering analysis plots from RNA-seq datasets were created using FPKM (fragments mapped per kilobase read per million times) values. The expression heatmap of the grape VvVAP27 gene family was drawn using TBtools.
Plant materials and treatments
-
The grapevine materials used in this study are Vitis vinifera 'Pinot Noir' and Vitis piasezkii 'Liuba-8', cultured in the Grape Repository of Northwest A&F University, Yangling, Shaanxi, China. The P. viticola population was collected from the susceptible Vitis plants as per previous studies[29−31]. Briefly, infected leaves were collected and washed in sterile distilled water three times. The leaves were positioned with the abaxial side facing up on sterile moist filter papers in trays and incubated overnight at room temperature to allow P. viticola sporulation. Leaves on which fresh sporangia developed were transferred into a large Petri dish and washed gently with sterile distilled water. The sporangium suspension was filtered with three-layer sterilized gauze. The concentration was adjusted to 5 × 104 sporangia/mL using a hemocytometer under a light microscope.
For inoculation, the third to fifth fully expanded leaves from the top were detached and washed three times in sterile distilled water, and inoculated with 10 μL drops of the sporangia suspension on the abaxial leaf surface. The inoculated leaves were placed on sterile Petri dishes (90 mm in diameter) containing three-layer sterile moist filter papers and incubated in an incubator at 23 ± 1 °C, 90% relative humidity, and a photoperiod of 16 h light and 8 h dark. Samples were collected at 0, 6, 12, 24, 48, 96, and 120 h post-inoculation (hpi), with 0 hpi as the control samples. The collected samples were promptly frozen in liquid nitrogen and stored at −80 °C. Each biological replicate was a pool of three independent leaves. The tobacco plant material, N. benthamiana, was routinely grown at 25/20 °C in a greenhouse under white light (18 h light/6 h dark).
RNA extraction and RT–qPCR
-
Total RNA was extracted from grapevine leaves using the RNA Mini Kit (Omega, USA) following the manufacturer's instructions. The EasyScript® One-Step gDNA Removal and cDNA Synthesis SuperMix kit (TransGen Biotech, China) was used to perform reverse transcription and synthesize double-stranded cDNAs. In the reverse transcription, 500 ng RNA was used with the Anchored Oligo (dT) 18 as the primer. The remaining reaction components included 10 μL of 2× ES Reaction Mix, 1.0 μL of gDNA Remover, 1.0 μL of EasyScript® RT Enzyme Mix, and sterile distilled H2O were added to reach a final volume of 20 μL. The reaction was carried out at 42 °C for 15 min, 85 °C for 10 s. Quantitative PCR was performed on an Applied Biosystems QuantStudio 6 (Thermo Fisher Scientific, USA) with PerfectStart® Green qPCR SuperMix (TransGen Biotech, China), according to the recommended protocol. In brief, each reaction mixture contained 10 μL of 2× TransStart Top Green qPCR SuperMix, 2.0 μL of cDNA template, 0.5 μL of each primer, and 8.0 μL of sterile distilled H2O. Cycling parameters included an initial step at 50 °C for 2 min and 94 °C for 30 s, followed by 45 cycles at 95 °C for 5 s, 59 °C for 15 s, and 72 °C for 30 s. Melt-curve analyses were performed with a program starting at 95 °C for 15 s and then a constant increase from 60 to 95 °C. Data were analyzed by the 2−ΔΔCT method for calculating gene relative expression levels with three biological replicates. Gene-specific primers were designed using Primer Premier 5.0 software and gene transcripts were normalized to VvActin as internal standards.
Transient transformation of Agrobacterium tumefaciens
-
The pCambia2300-VAP27s-GFP construct was introduced into the A. tumefaciens GV3101 strain, and recombinant colonies were verified through growth on a selective medium and PCR analysis. For Agrobacterium-mediated transient transformation assays, bacterial cells were collected by centrifugation and then resuspended in infiltration buffer (10 mM MgCl2, 10 mM MES, 200 μM acetosyringone). The bacterial suspension was diluted to a concentration of OD600 = 0.6 and incubated for 3 h at 28 °C before infiltration. Infiltration on tobacco leaves was carried out using a syringe without a needle.
Location analysis
-
Laser confocal microscopy was employed to determine the localization of the VAP27 gene family in plants. The VAP27 gene was inserted into the pCambia2300-GFP vector and the recombinant plasmid was confirmed by Sanger sequencing. Then the recombinant plasmid was transformed into the Agrobacterium strain GV3101. The monoclonal plaque was amplified in liquid culture and confirmed by PCR analysis. For visualization, the ER-RK marker was co-injected with VAP27-GFP into the N. benthamiana leaves. The transformed N. benthamiana leaves were observed using confocal microscopy (TCS SP8 of Leica). The excitation wavelength for green fluorescent protein was set to 488 nm.
Phytophthora capsici inoculation
-
The mycelium of P. capsici was first cultivated on 10% V8 juice agar medium and then was transferred into 10% liquid V8 medium and cultured in 25 °C darkness for 5 d. The developed hyphae were collected and resuspended in sterile water at 4 °C for 30 min, followed by incubation at room temperature for 30 min to allow the release of zoospores from sporangia. The resulting sporangial suspension was adjusted to 1.0 × 104 sporangia/mL.
For the P. capsica infection experiment, the A. tumefaciens containing certain plasmids were injected into N. benthamiana. At 48 h post-infection, the inoculated leaves were detached, and their petioles were first wrapped in sterile cotton and then wrapped in two layers of sterile wet filter paper. Then the leaves were first treated using 0.1% Tween-20, followed by inoculation with 30 μL zoospore suspension of P. capsica. The infected leaf samples were kept in the dark at 25 °C to allow the P. capsica development. The lesion area was statistically analyzed.
-
A total of 12 VAP27 genes in the grape genome were identified, designated as Vitis vinifera VAP27 (VvVAP27)1−12 according to their chromosomal positions (Table 1). The genomic distribution revealed an uneven mapping of VAP27s on eight out of the 19 grape chromosomes. Specifically, VvVAP27-1, VvVAP27-2, VvVAP27-3, VvVAP27-4, VvVAP27-7, and VvVAP27-10 were located on Chromosome 2, Chromosome 5, Chromosome 8, Chromosome 12, Chromosome 14, and Chromosome 19, respectively. VvVAP27-5 and VvVAP27-6 were situated on Chromosome 13, while VvVAP27-8 and VvVAP27-9 were positioned on Chromosome 15. VvVAP27-11 and VvVAP27-12 were putatively located on the 'Chromosome Unknown'. Further study will delve into unraveling the biological functions of these 12 VAP27 genes.
Table 1. Chromosome distribution of identified 12 grapevine VAP27 genes. Detailed information, including gene locus, gene symbol, length, chromosome, and site is available in the Ensembl Plants Database.
Protein name Gene ID Chr Length (aa) Annotation VvVAP27-1 Vitvi02g00545 2 238 PREDICTED: vesicle-associated protein 1-1 VvVAP27-2 Vitvi05g00360 5 293 PREDICTED: vesicle-associated protein 1-2 VvVAP27-3 Vitvi08g00137 8 532 PREDICTED: ankyrin-1 VvVAP27-4 Vitvi12g00638 12 259 PREDICTED: vesicle-associated protein 4-1 VvVAP27-5 Vitvi13g00099 13 470 PREDICTED: ankyrin repeat, PH and SEC7 domain containing protein secG VvVAP27-6 Vitvi13g00850 13 136 PREDICTED: hypothetical protein VITISV_015240 VvVAP27-7 Vitvi14g00347 14 336 PREDICTED: vesicle-associated protein 2-2 VvVAP27-8 Vitvi15g00677 15 239 PREDICTED: vesicle-associated protein 1-2 VvVAP27-9 Vitvi15g00713 15 239 PREDICTED: vesicle-associated protein 1-3 VvVAP27-10 Vitvi19g00304 19 264 PREDICTED: vesicle-associated protein 4-1 VvVAP27-11 Vitvi10g04245 Un 264 PREDICTED: vesicle-associated protein 4-2 VvVAP27-12 Vitvi00g04146 Un 348 PREDICTED: LOW QUALITY PROTEIN: vesicle-associated protein Phylogenetic analysis of the VAP27 gene family
-
To elucidate the evolutionary relationships within the VAP27 gene family, we conducted a comprehensive analysis involving 53 VAPs, including 10 from Arabidopsis, 17 from rice, 15 from tomato, and 12 from grape, and the result was visualized by constructing a phylogenetic tree (Fig. 1). The 53 VAP27 members across these four species fell into three distinct groups (Fig. 1 Clade I−III). Clade I, consisted of VvVAP27-1, VvVAP27-6, VvVAP27-8 and VvVAP27-9 gene. Clade II contained 3 VvVAP27 members: VvVAP27-2, VvVAP27-7 and VvVAP27-12. Clade III emerged as the most populated, encompassing five VvVAP27 members: VvVAP27-3, VvVAP27-4, VvVAP27-5, VvVAP27-10, and VvVAP27-11.
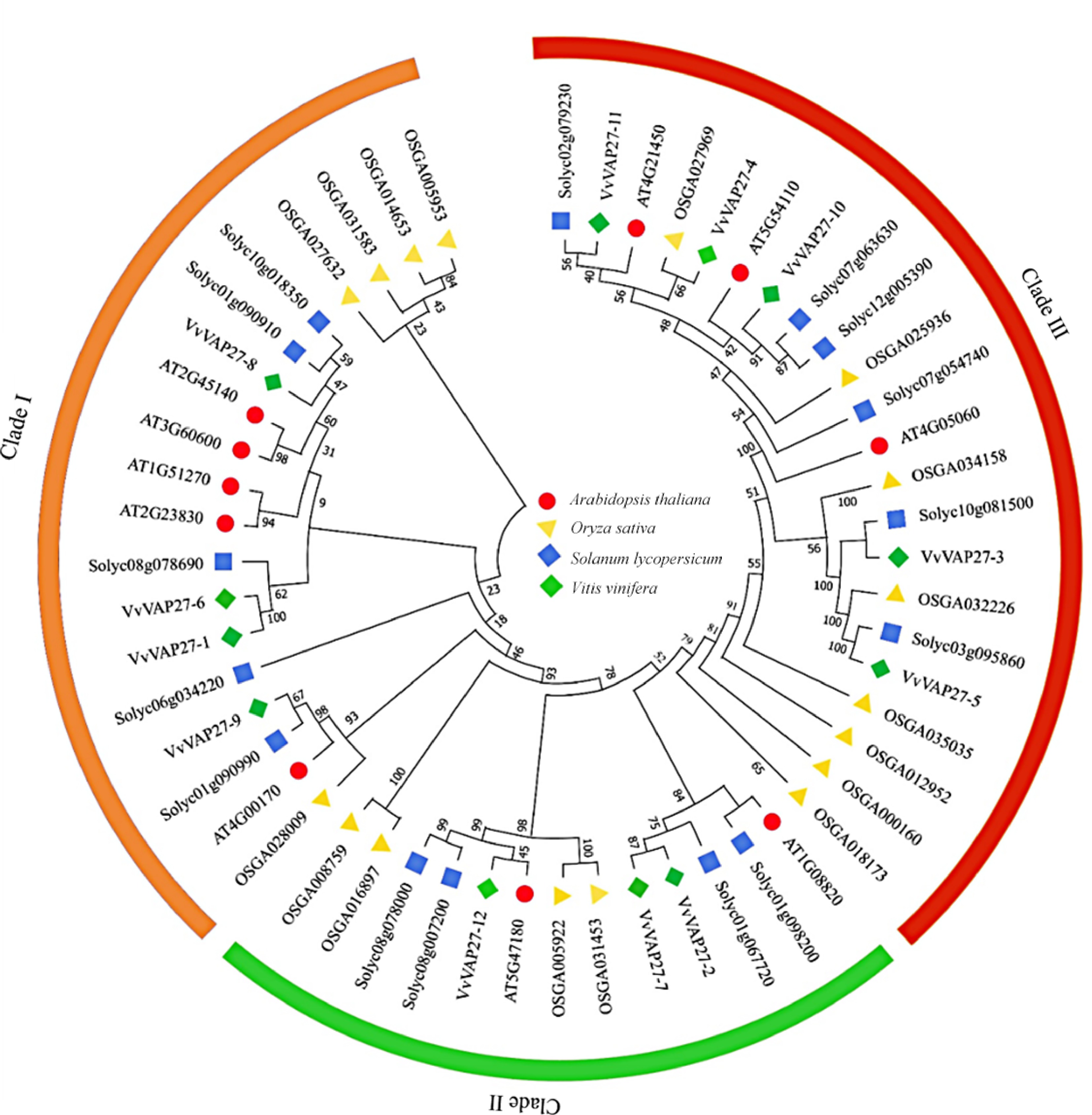
Figure 1.
Unrooted phylogenetic tree of VAP27s in grape, Arabidopsis, rice, and tomato. Vv: Vitis vinifera L. grape species; AT: Arabidopsis thaliana; OS: Oryza sativa; Soly: Solanum lycopersicum.
The clustering patterns suggest a closer evolutionary proximity of the VAP family in grapes to that of dicotyledon tomatoes compared to rice. This supports the reliability of the analysis results.
Structural analysis of grape VAP27 genes
-
The gene structures of the 12 grapevine VAP27s were explored through a comprehensive examination of exon/intron boundaries. The varying length and splicing patterns observed among the 12 VAP27s are depicted in Fig. 2a. The structural analysis showed a range of intron numbers from 1 to 7. Notably, VvVAP27-3, VvVAP27-6, and VvVAP27-7 were absent of introns, while VvVAP27-5 exhibited a singular intron. The remaining VAP27 member's genes had between six and seven introns (Fig. 2a). The results revealed significant diversity within the VAP27 family.
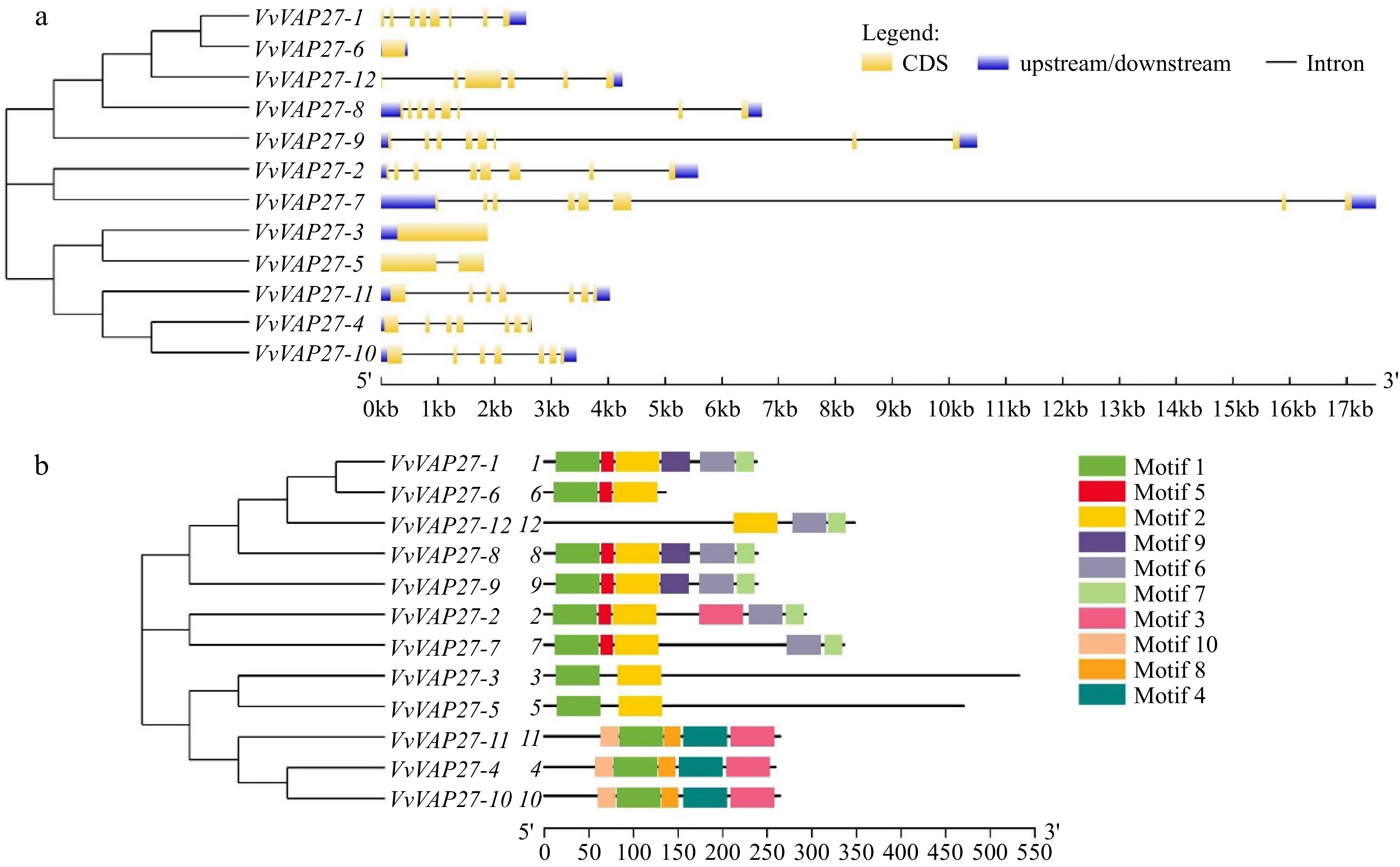
Figure 2.
(a) Intron-exon structure and (b) conserved motifs of VvVAP27.
According to previous studies, motifs recognized as playing an important role in interaction and signal transduction within the transcriptional complex[32,33] were analyzed using MEME for the 12 conservative VAP27 genes. This was conducted also because these motifs are closely related to gene classification. Among these VAP27s, a total of 10 motifs were identified (Fig. 2b), with Motif 1 present in all 11 members except VAP27-12, which indicates its high conservation within the VAP27 gene family. Motif 8 and Motif 9 were the least conserved, found only in VvVAP27-4, VvVAP27-10, and VvVAP27-11 (Motif 8), and VvVAP27-1, VvVAP27-8, and VvVAP27-9 (Motif 9). The high sequence similarity among genes within the same branch suggests shared functions and roles in plants. The analysis of motif and gene structure analysis enrich our understanding of the VAP27 family's classification, providing a robust theoretical foundation.
Expression profiles of VAP27s in different tissues
-
VAP27 RNAi induces various defects in plant morphology, pollen, seed, and root development in Arabidopsis[1], we anticipated a similar involvement of the VvVAP27 gene in grape growth and development. To investigate this, we examined the expression profiles of the 12 VAP27 genes across different tissues of grapevine (Fig. 3). These tissues represented distinct growth and development stages of grapevine, including root, young stem, leaf, inflorescence, skin, veraison berry, and tendril. Examination of transcriptome data from the VvVAP27 family revealed significant variations across different tissues. The majority of family members (VvVAP27-1 to VvVAP27-10) exhibited comparable expression levels in tissues including flowers, berries, leaves, stems, seeds, and shoots, suggesting their involvement throughout various stages of plant growth and development. Only a subset of genes (VvVAP27-11 and VvVAP27-12) showed significant differences in expression among tissues. Expression of the VvVAP27-11 gene was higher in the berries than in the other tissues that maintained relatively consistent levels. In contrast, VvVAP27-12 showed transcriptional peaks exclusively in seeds and flowers, suggesting a potential association of VvVAP27-12 with flowering and fruit development.
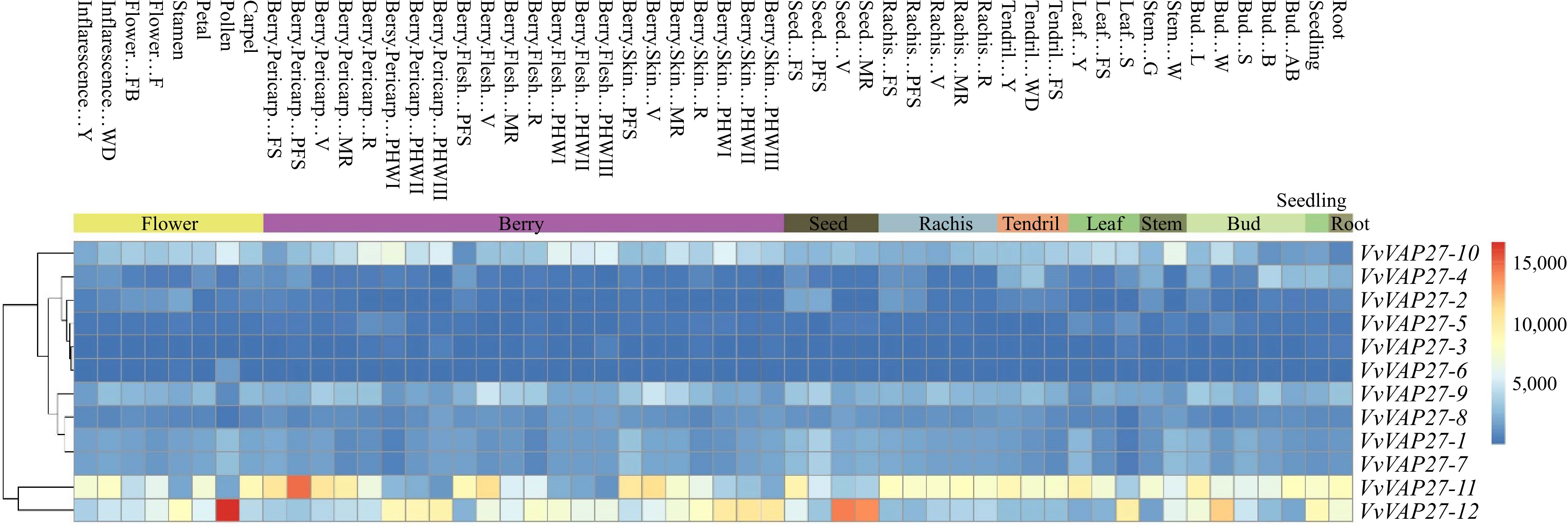
Figure 3.
Expression profiles of the grape VvVAP27s gene. Hierarchical clustering of expression profiles of grape VAP27 genes across different tissues.
Expression patterns of VAP27 genes post-plasmopara viticola inoculation
-
It has been documented that the VAP27 gene family is involved in regulating plant disease resistance against external pathogen infection[6,18−22]. Therefore, we explored whether the VAP27 family exhibits similar functionality in grapevine downy mildew resistance. Our investigation focused on the expression levels of 12 VAP27 gene members at eight time points post-downy mildew inoculation (0, 6, 12, 24, 48, 72, 96, and 120 hpi). Utilizing RT-qPCR, we assessed whether the VAP27 gene responded to the induction of Grape downy mildew (Fig. 4). Vvactin1 was used as a grapevine internal reference gene for normalization[25].
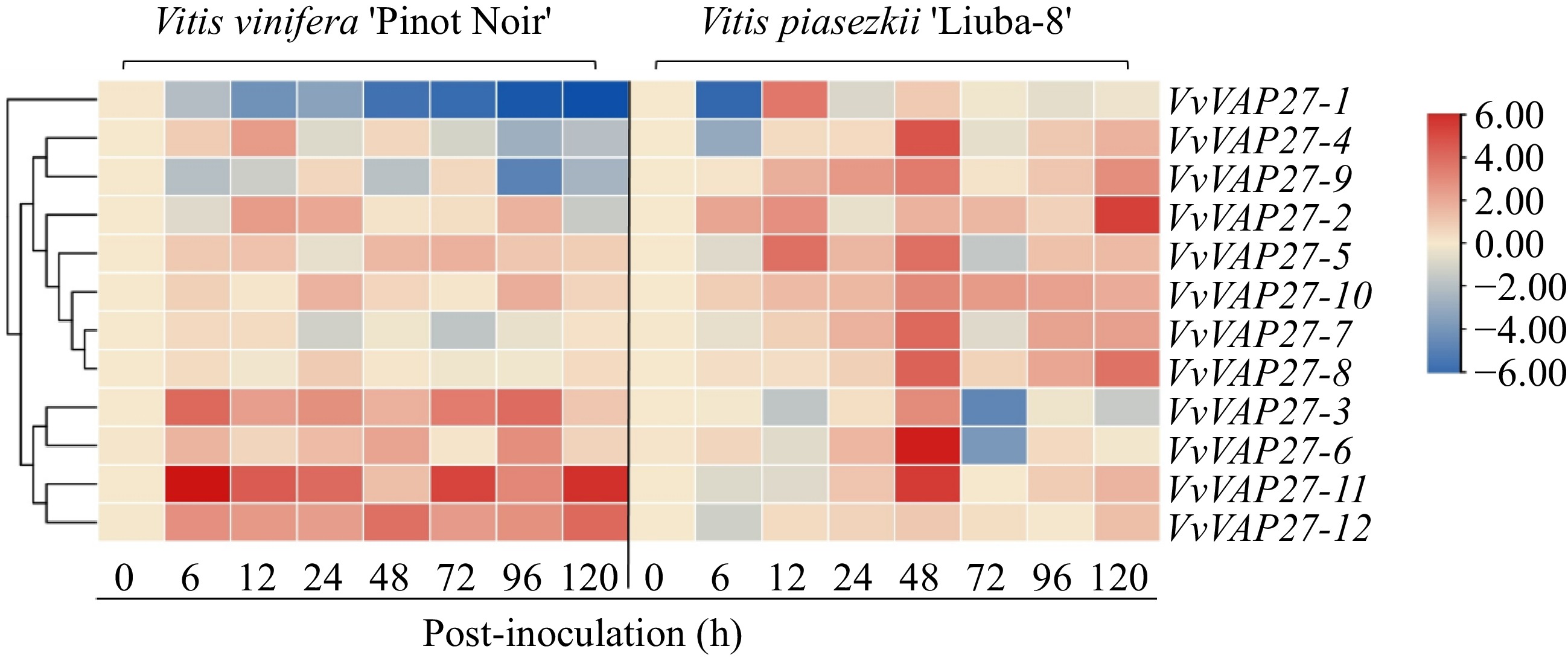
Figure 4.
Heat map showing the expression profiles of VvVAP27 genes at different time points post-downy mildew infection. The color scale represents expression levels, with red indicating high expression level and blue indicating low expression level. The expression was normalized and the data are displayed as log2 values.
We discovered that genes in the VvVAP27 family genes exhibited specificity in responses to downy mildew induction (Fig. 4). There were a few members significantly overexpressed at the early stage of downy mildew infection in 'Liuba-8'. Only VvVAP27-6 had the highest expression level at 48 hpi compared to other time points. VvVAP27-2 was highly expressed at 120 hpi, with no difference found at other time points. VvVAP27-3, VvVAP27-6, VvVAP27-11, and VvVAP27-12 genes were highly expressed throughout the downy mildew infection period in 'Pinot Noir'. This indicated that these four genes were positively responsive to the induction of downy mildew, suggesting an important role in the grapevine's defense against downy mildew invasion. The expression levels of the other eight genes remained unchanged across different infection periods. We postulate that the varying expression patterns among different members may be related to the regulation of VAP27-mediated plant disease resistance, possibly involving distinct mechanisms of immunity. However, further experimental verification is needed to substantiate these hypotheses.
Subcellular localization of VvVAP27 genes
-
To better explore the function of the VvVAP27 gene family, subcellular localization analysis was conducted on some selected VvVAP27 genes. By detecting GFP-tagged proteins, we found that most of the genes were localized to the endoplasmic reticulum (ER). This localization aligns with previous literature reports indicating that membrane proteins of vesicle-associated proteins function by participating in the formation and regulation of plant cell membranes. The endoplasmic reticulum participates in the formation of cell membranes and is closely related to secretory vesicles that function in transporting secretory proteins to various parts of the plant to contribute to plant growth, development, and disease resistance[34,35]. VvVAP27-2, VvVAP27-4, VvVAP27-6, and VvVAP27-9 are all mapped to the endoplasmic reticulum. However, VvVAP27-2, VvVAP27-4, and VvVAP27-6 are also detected in the nucleus in addition to the ER (Fig. 5). Based on the analysis of the expression pattern induced by downy mildew and the Phytophthora capsici infection experiment, we speculate that the subcellular localization may affect the gene expression.
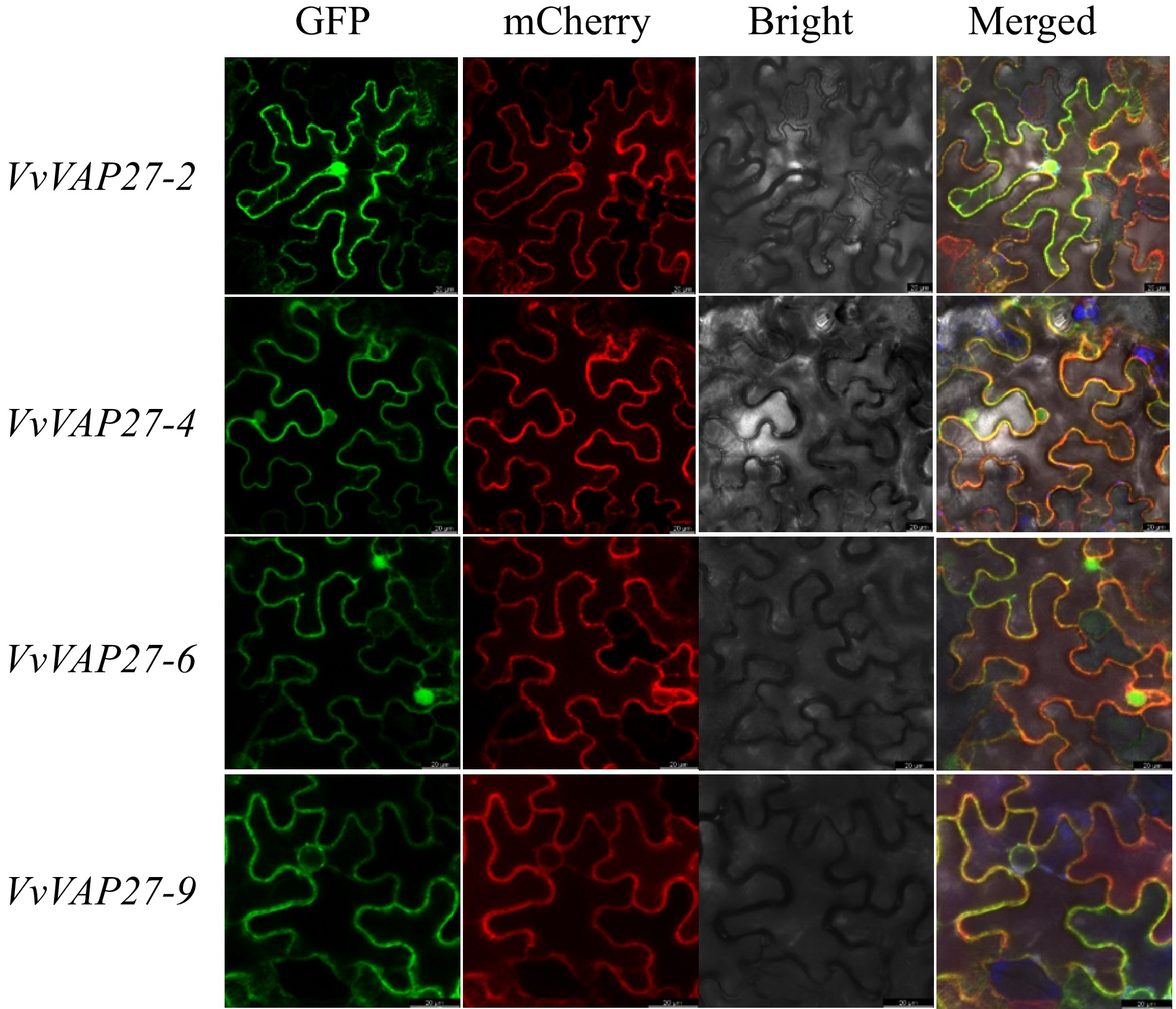
Figure 5.
Subcellular localization analysis of VvVAP27 members.
Response of VAP27 gene to P. capsici inoculation
-
To further study the role of VAP27 genes in disease resistance, the VvVAP27 genes that were induced by grape downy mildew in tobacco leaves were screened. These tobacco leaves were transiently transformed by A. tumefaciens that carry a high-level expression vector with an individual VvVAP27 gene insert before being inoculated with P.capsici spore suspension. The findings revealed that VAP27 gene members inhibited the occurrence of the pathogenicity and significantly enhanced the resistance of tobacco leaves to the pathogen. However, the efficacy of pathogen inhibition varied among different VAP27 members. This result is consistent with previous studies on gene responses to downy mildew infection.
Specifically, both VvVAP27-6 and VvVAP27-9 exhibited a consistent phenotype, inhibiting infection by pathogens (Fig. 6a). Leaf lesion areas were smaller in VvVAP27-6 and VvVAP27-9 expressing leaves compared with controls, suggesting that they effectively promoted plant immunity (Fig. 6b) and that VvVAP27-6 had a higher inhibitory capacity than VvVAP27-9. VvVAP27-2 and VvVAP27-4, did not differ significantly in the size of the lesion area compared with empty-carrier controls. The heterogeneous functions within this family underscore the need for further experimental studies to elucidate the roles of the remaining genes.
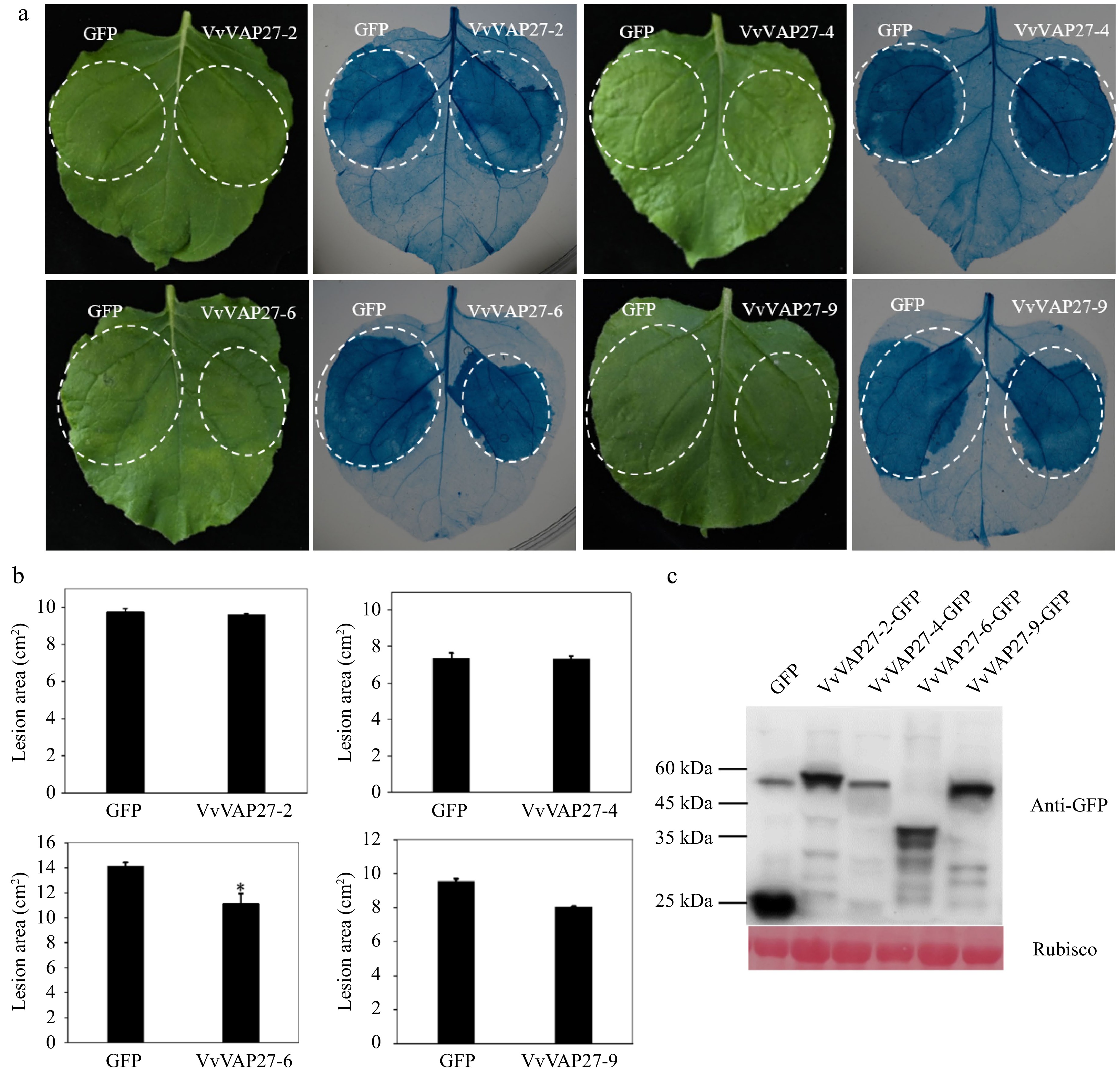
Figure 6.
Responses of VvVAP27 induced by Phytophthora capsici infection. The inoculation of Phytophthora capsici was conducted on Nicotiana benthamiana leaves transiently transformed with VvVAP27. (a) Observation of the phenotypes of VvVAP27 family members and the control after inoculation with Phytophthora capsici, visualized by trypan blue staining. (b) Statistical analysis of the lesion areas caused by Phytophthora capsici infection on Nicotiana benthamiana leaves transiently transformed by VvVAP27. (c) The expression of VvVAP27 family members and GFP protein was detected by Western blot. The experiment was repeated three times and asterisks represent the level of significant differences (* p < 0.05, ** p < 0.01).
-
Vesicle-associated membrane proteins (VAMP-associated proteins) (VAPs) are a family of proteins widely expressed in plants, which play a key role in plant defense against both biotic and abiotic stresses. In this study, a family of 12 VAP27 genes were identified in grapes using bioinformatic methods. Consistent with previous studies, the VAMP gene family has demonstrated multifaceted involvement in diverse defense processes across different plant tissues. VAP27-1 and VAP27-3, as non-plant VAP homologs[5], have been localized extensively to the ER and EPCS[1,5,16]. These proteins were identified to promote plant endocytosis and play a role in endocytosis. In Arabidopsis thaliana, VAMP721/722 have been identified as essential factors for growth, cell division, and responses to abiotic stress[18−22]. The PEN1-SNAP33-VAMP721/722 pathway in Arabidopsis thaliana facilitates the transport of vital materials for cell viability to the endoplasmic reticulum[36,37]; VAMP721 and VAMP722 are involved in the secretory transport of substances to endosomal compartments of the plasma membrane to promote the formation of cell plates during plant cytokinesis[6]. The phylogenetic tree analysis revealed a notable similarity in the quantity and gene structure of the VAP27 gene family between grape, Arabidopsis thaliana, and tomato. Hence, it can be inferred that the VAP27 gene family in grape shares similar functions with those in Arabidopsis thaliana[36], and plays comparable roles in growth, development, and immune resistance mechanisms. Additionally, we analyzed the gene structure of 12 identified VvVAP27 genes using MEME. Results (Fig. 2) showed that most of the VvVAP27 genes (VvVAP27-1, VvVAP27-2, VvVAP27-3, VvVAP27-6, VvVAP27-7, VvVAP27-8, VvVAP27-9) contain conserved domains, especially VvVAP27-3 and VvVAP27-5; VvVAP27-1, VvVAP27-8 and VvVAP27-9; VvVAP27-4, VvVAP27-10 and VvVAP27-11 placed within the same group in the phylogenetic tree classification (Fig. 1). This observation may explain the specific biological functions associated with each subfamily. The analysis of motif and gene structure provides a further theoretical basis for the classification of the VvVAP27 subfamily, guiding subsequent in-depth functional studies within these identified groups.
To understand the regulatory mechanisms of the VAP27 gene family on the growth and development of grapevine, we analyzed the regulatory effect of the VvVAP27 gene family on the growth and development of grapevine using transcriptome content assay (Fig. 3) across different grape tissues (flowers, seeds, leaves, buds, berries, tendrils, stems, roots). Our transcriptome data analysis showed the highest expression of VvVAP27-11 in berries than in other tissues, implying its potential role in regulating fruit development and quality and promoting fruit setting. Similarly, the expression levels of VvVAP27-12 were higher in flower and seed than in other tissues, indicating that VvVAP27-12 plays an important role in flower induction, seed setting, and growth regulation. This study provides a theoretical basis for further understanding the function of the VAP27 family members in the process of grape growth and development.
It has been found that the VAP gene family can induce plant cell autonomous immunity, acting on the cell surface or post-pathogen entry, thereby impeding pathogenesis. This phenomenon has been well-documented in various plant species, including tomato, Arabidopsis, and tobacco. In tobacco, VAPB proteins interact with proteins in the intestine of tobacco whitefly (Bemisia tabaci) during the transmission of tomato yellow leaf curl virus (TYLCV), and silencing VAPB results in an increase in virus number and transmission rate, demonstrating that VAPB can play a key role in resistance to TYLCV[4]. Moreover, in Arabidopsis, the SNARE proteins VAMP 721/722 direct secretory vesicles to pathogen-attack sites during immune responses, indicating that these vesicles deliver immune molecules and function in immune responses[36]. Additionally, SYP132, an essential protein for defense against bacterial pathogens, specifically interacts with VAMP721/722 in response to the immune control of P. syringae[21]. Given these findings, we were intrigued by the possibility that the VAP27 gene family might play a similar role in disease resistance in grapes. Grapevine downy mildew, caused by the oomycete P. viticola, is one of the most serious diseases in grape production. P. viticola was originally endemic to North America, but it has now spread to all major grape-producing regions worldwide[37]. This study revealed that the majority of VvVAP27 members exhibited responses to downy mildew infection. Notably, VvVAP27-1, VvVAP27-2, VvVAP27-4, and VvVAP27-6 were significantly up-regulated at 0, 6, and 12 h after infection, and VvVAP27-12 was highly expressed at 24, 48, and 72 h after infection with P. viticola, while other genes were highly expressed only at specific stages. These findings suggest that members of the VAP27 gene family are likely to respond to downy mildew infection. Our results provide a new idea to study the effect of the VAP27 gene on grapevine downy mildew, but further research is needed to study the mechanisms of VAP27 gene action.
In this study, we identified a new grape gene family, named the VAP27 gene family. Through bioinformatic analysis and transcriptome sequencing, we uncovered striking structural and functional similarities between the VAP27 gene family in grape and those in Arabidopsis thaliana and tomato. The current findings suggest a potentially significant role for this gene family in the growth and development of the grape, as well as in orchestrating immune responses against downy mildew.
-
The vesicle-associated protein-membrane protein gene family (VAP27) in the grape genome was identified, which consists of 12 gene members. Within this family, some members exhibit localization on the endoplasmic reticulum, and a minority reside within the nucleus. The present results demonstrate the induction of the gene family in response to downy mildew in grape and their ability to inhibit the infection of P. capsici, thus playing an important role in plant disease resistance.
-
The authors confirm contribution to the paper as follows: conceptualization, writing – review & editing: Xu Y, Liu G; validation, visualization, writing – original draft: Li R, Wang B; investigation: Li R, Wang B, Zha M, Zhang K, Li M, Xie L, Chen X, Liu G; funding acquisition: Liu G. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article.
This work was supported by grants from the National Natural Science Foundation of China (31972374, 32372660, and 31601716).
-
The authors declare that they have no conflict of interest. Yan Xu is the Editorial Board member of Vegetable Research who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and the research groups.
-
# Authors contributed equally: Ruonan Li, Bianbian Wang
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Li R, Wang B, Zha M, Zhang K, Li M, et al. 2024. Identification and characterization of grape VAP27 gene family and their roles in disease resistance. Fruit Research 4: e019 doi: 10.48130/frures-0024-0019 

Identification and characterization of grape VAP27 gene family and their roles in disease resistance
- Received: 24 January 2024
- Revised: 03 April 2024
- Accepted: 24 April 2024
- Published online: 17 May 2024
Abstract: Vesicle-associated membrane protein (VAMP)-associated proteins (VAP27s), which are widely expressed in plants and animals, play an important role in metabolism, physiology, growth, and development, disease resistance, and immunity. While the function of this family has been elucidated in model plants like Arabidopsis thaliana and tomato, its role in grapevine remains unclear. In this present study, 12 vesicle-associated protein-membrane protein genes were identified in the grapevine genome by bioinformatics, designated as the VAP27 gene family. A phylogenetic tree, encompassing 53 genes from three model plants, Arabidopsis thaliana, Oryza sativa, and Solanum lycopersicum, revealed the subdivision of the VAP27 gene family into three subfamilies, each presumably serving different functions, besides localizing in endoplasmic reticulum, individual members also localize in nucleus. Additionally, we compared the transcriptional levels and subcellular localizations of the VvVAP27 family members across different plant tissues (flower, leaf, seed, root, fruit, tendril, and stem), indicating site-specific functionalities for different gene members. To investigate the responsiveness of the VAP27 gene family to pathogen infection, particularly Plasmopara viticola on host plants, we analyzed the expression patterns of VAP27 genes post-infection. Our findings revealed divergent expression profiles among different members at different stages of infection. The gene family responded to the infection of downy mildew on grapevine and could inhibit the spread of Phytophthora capsici lesions in Nicotiana benthamiana. These results provide an important basis for further studies delving into the functions of the VAP27 gene family in plant growth and disease resistance.
-
Key words:
- Grapevine /
- VAP27 /
- Grape downy mildew /
- Disease resistance