









-
Tea (Camellia sinensis (L.) O. Kuntze) stands as China’s earliest documented tree crop, boasting a domestication history spanning over 3,000 years. Initially employed as a medicinal herb with roots dating back nearly 5,000 years, it later evolved into a beverage widely embraced for consumption[1]. On a global scale, cultivated tea plants are classified into two primary groups: C. sinensis var. sinensis (CSS) and C. sinensis var. assamica (CSA)[2].
Hainan Island, positioned in the northern part of the South China Sea, has a rich history of tea plant cultivation and extensive planting areas. There were reports of the abundant tea plant resources on Hainan Island at the end of the Qing Dynasty. For instance, the American missionary and botanist Benjamin Couch Henry uncovered a significant number of wild tea trees during his extensive exploration of the Li ethnic group area in Hainan, confirming the abundance of ancient tea tree resources on the island[3]. As the Yunnan-Guizhou Plateau is widely recognized as a potential geographical origin of tea[4−6], most studies on tea plant population genomics encompass samples from southwestern China, particularly CSA varieties[1, 6−8], leaving research on tea plants in Hainan Island relatively sparse. The self-incompatibility of tea trees results in high offspring heterozygosity, and the abundant wild tea plant germplasm on the island provides a wealth of genetic variation, laying the groundwork for cultivating new varieties with desirable traits[7]. Despite Hainan Island’s abundance of tea resources, fully comprehending the genetic resources of tea plants there poses a challenge due to its unique climate and geographical environment. Hence, a genome-wide investigation into the genetic diversity of Hainan tea is imperative for a comprehensive understanding of the genetic resource background of Hainan tea.
It is noteworthy that the tea plant species on the island closely resembles CSA and is referred to as ‘Hainan dayezhong’[9]. However, evidence is insufficient to conclusively determine whether the Hainan dayezhong belongs to CSA or not. The classification of Hainan tea presents a significant challenge for several reasons: Firstly, C. sinensis plants are prone to hybridization between different species, posing a challenge in accurately classifying various hybrid progenies. Secondly, numerous morphological characteristics of tea plants resemble each other, complicating precise taxonomic delineations[10]. Lastly, traditional classification of tea plants primarily relies on morphological characteristics, which may sometimes conflict with the latest molecular-based classification results[8]. Despite Hainan tea’s identification by the National Crop Variety Approval Committee in 1985, its taxonomic status within the Camellia genus on Hainan Island remains unclear due to the absence of support from modern genomic research data.
Islands, as an ideal system for studying the effects of geographical isolation and long-distance diffusion, offer valuable insights into species evolution, encompassing phenomena such as adaptive radiation and speciation[11]. Previous studies have documented the discovery of several new plant species on Hainan Island, including Holttumochloa[12], Euphorbia[13], Cycadaceae[14], among others. Moreover, advancements in whole-genome resequencing technology have confirmed the independent evolutionary histories and parallel domestication processes of CSS and CSA[7, 8]. Building upon these findings, our hypothesis suggests that tea trees on Hainan Island may constitute a distinct species separated from CSS and CSA, and that Hainan tea has undergone an independent evolutionary trajectory on the island.
To furnish molecular evidence regarding the genomic divergence and relationship of Hainan tea with CSS and CSA, and to elucidate the genetic background of Hainan tea on Hainan Island, we procured 500 samples of Hainan tea from the Baisha, Qiongzhong, Wuzhishan, and Ledong regions of Hainan Province, China. Employing whole-genome resequencing technology, we identified SNPs in the Hainan tea samples and constructed a phylogenetic tree that included both cultivated tea and Hainan tea, utilizing the Yunkang 10 as the reference genome. Subsequently, detailed analyses of population structure and kinship relationships were conducted to offer a comprehensive understanding of the population structure and genetic diversity of Hainan tea and to unveil the phylogenetic relationships between Hainan tea and global Camellia sinensis varieties. This study furnished robust genomic data support and further corroborated the independent status of Hainan tea within the taxonomy of Camellia sinensis. Concurrently, these findings furnished a crucial scientific foundation for the conservation of tea germplasm resources and molecular breeding on Hainan Island. Furthermore, the research methodologies and techniques employed herein hold the potential to provide valuable insights into the origin and domestication analyses of other species on Hainan Island, as well as for the investigation of genetic diversity.
-
In this study, 500 tea tree samples were collected from four major tea-producing regions in Hainan: Ledong, Qiongzhong, Baisha, and Wuzhishan (Fig. 1). Notably, the samples encompassed a substantial number of ancient tea trees. Detailed sample information is provided in the Methods and Materials section and Supplemental Table S1. Following the sequencing, a total of 6.9 Tb of raw sequencing data were obtained. Subsequently, the original data source underwent filtration and was aligned with the reference genome (Yunkang 10), yielding a final average alignment rate of 98.98%. Notably, A-MCXB3-1D was excluded from the dataset due to its notably low mapping rate of 27.27% (Supplemental Fig. S1; Table S1).
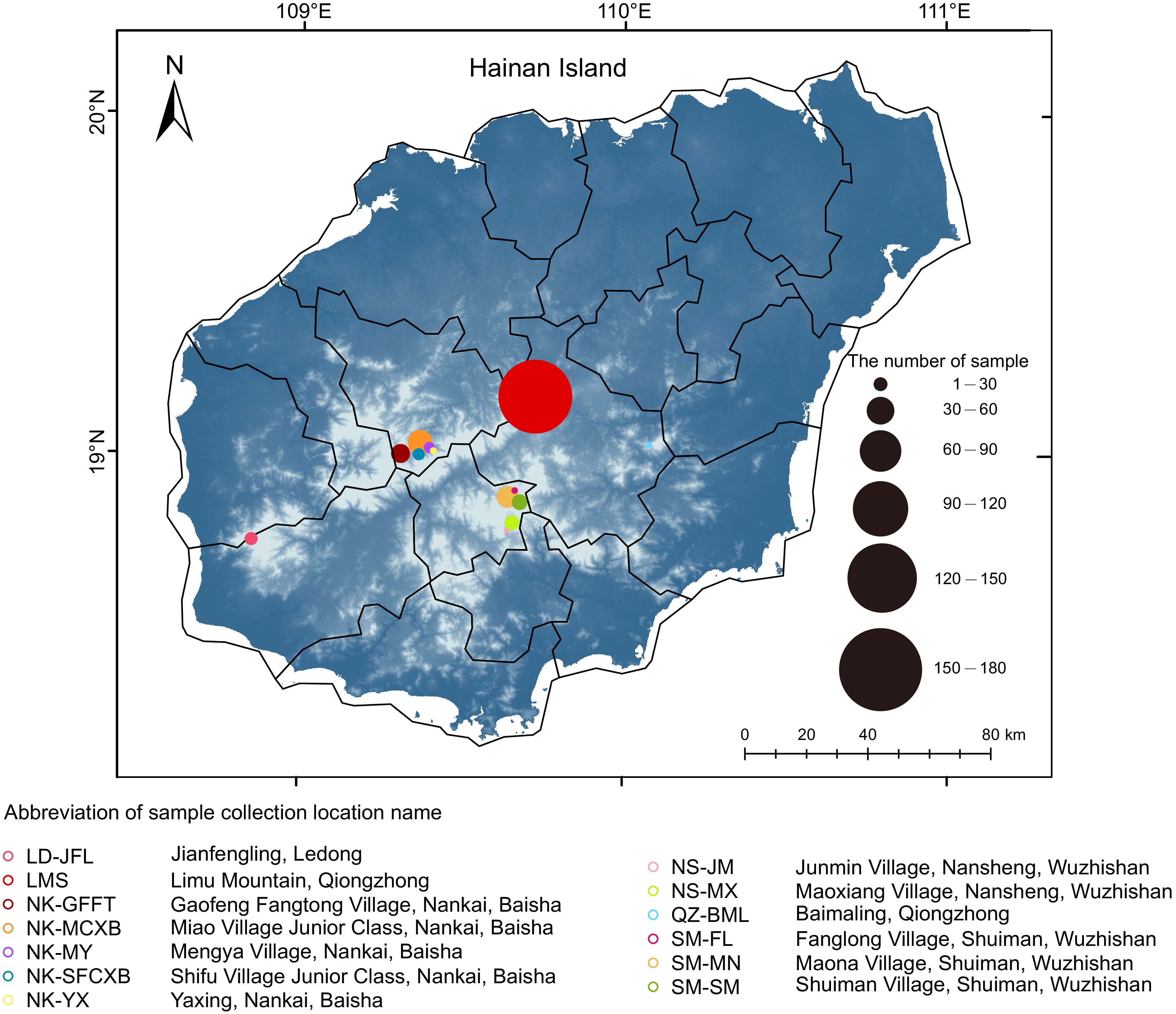
Figure 1.
Geographical distribution of tea samples collected and analyzed in this study.
The color of circles denotes the collection area of Hainan tea samples, with circle size indicating the corresponding sample count. Larger circles indicate higher sample counts in the respective areas.Based on the results from Supplemental Fig. S2, which contain information about SNPs initially detected using GATK, hard filtering conditions were established initially. Subsequently, SNPs with a minor allele frequency (MAF) of at least 0.05 were retained, resulting in a total of 32,334,340 SNPs (Supplemental Table S2). Among them, 91.3% were located in the intergene region, 4.85% in the intron region, 0.12% in the 5' UTR, and 0.28% in the 3' UTR. In addition, 1.01% of SNPs were located in the upstream region of the gene, 1.06% in the downstream region. Exon SNPs accounted for 1.34% of the total SNPs, of which non-synonymous SNPs and synonymous SNPs accounted for 49.82% and 47.93%, respectively (Tables 1, 2).
Table 1. The number of SNPs in different genome structures.
Variants Type Core set SNP Total 32,334,340 Intergenic 29,520,274 Intronic 1,566,641 Exonic 433,604 5' UTR 40,383 3' UTR 92,101 UTR5;UTR3 229 Upstream 326,710 Downstream 341,541 Upstream;downstream 7,803 Splicing 4,838 Exonic;splicing 216 Table 2. The number of large-effect SNPs.
Variants Type Core set SNP Total (exonic + exonic;splicing) 433,820 Nonsynonymous 243,634 Synonymous 179,902 Nonsyn/Syn ratio 1.35 Stop-gain 9,699 Stop-loss 518 Unknown 67 Phylogenetic analysis
-
To explore the phylogenetic relationship between tea trees of CSS and CSA on Hainan Island, genomic data of CSS and CSA were obtained from various global regions from the teabase database (
http://teabase.ynau.edu.cn/ )[15], as well as from the Genome Sequence Archive project number PRJCA001158[8]. Additionally, KM6 (Camellia cuspidata) was selected as an outgroup. The chosen tea plant materials are listed in Supplemental Table S3. Subsequently, we constructed a phylogenetic tree between the resequenced samples and globally cultivated tea trees using the maximum likelihood method with SNPs extracted from prior results. Phylogenetic analysis revealed distinct categorization of the samples into four main classes: global Aassamica1, global Assamica2, global Sinensis, and tea trees from Hainan Island. Notably, global Assamica and Sinensis clustered together on one side of the tree, while the tea tree samples from Hainan Island formed a separate, distinct cluster, without mingling with global Assamica and Sinensis. Particularly noteworthy is the significant geographic clustering observed in samples collected from the Limu Mountain area (abbreviated as LMS in Fig. 2), forming a subgroup within the Hainan tea sample cluster. Conversely, samples from other regions lacked a discernible pattern of geographic or regional clustering. For simplicity, we collectively refer to Hainan tea samples as ‘Hainan tea’. Based on the phylogenetic tree clustering results, we abbreviated tea samples from the Limu Mountain region as LMS and those from outside the Limu Mountain region as OLMS.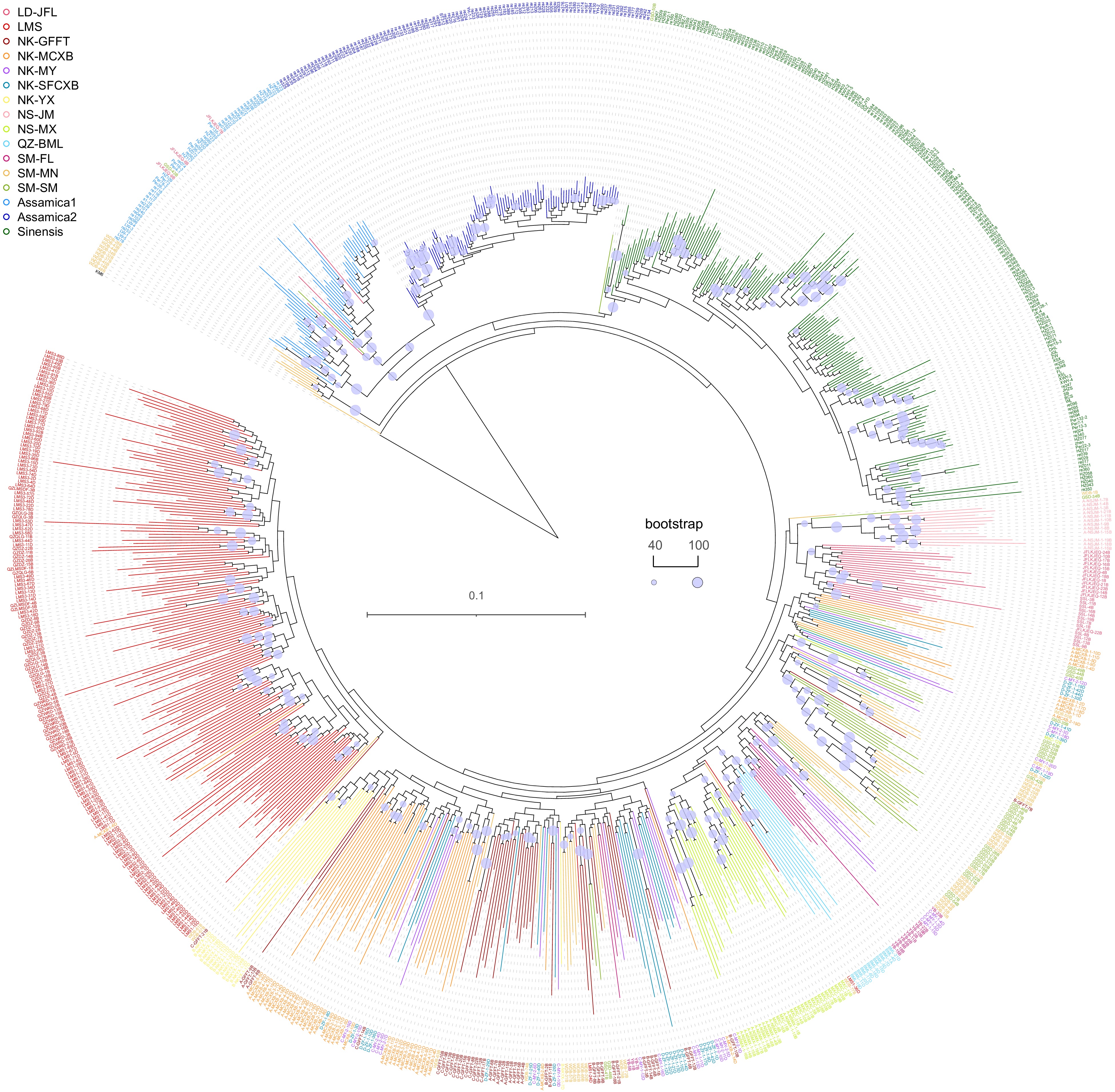
Figure 2.
Phylogenetic relationship between Hainan tea and cultivated tea.
Branch color indicates the sample source. Blue, purple, and green branches in the upper section of the phylogenetic tree denote cultivated tea across global regions. CSA is divided into Assamac1 (blue) and Assamac2 (purple). The lower section illustrates the phylogenetic relationships among Hainan tea samples from distinct regions.Further population rooted tree analysis (Supplemental Fig. S3) indicated that Hainan tea exhibited significant genetic divergence from CSS and CSA, making it challenging to classify them within the same category. This suggested a considerable evolutionary divergence of Hainan tea from CSS and CSA, although additional evidence is required to bolster this hypothesis. Notably, a discernible genetic distance exists between OLMS and LMS in the evolutionary tree, and they do not form a distinct category, consistent with the findings of phylogenetic analysis. In comparison to the outgroups, Hainan tea clustered more closely with global Assamica and Sinensis, prompting speculation that Hainan tea may belong to a Camellia species distinct from CSS and CSA.
Population structure and principal component analysis (PCA)
-
Utilizing the data from Fig. 2 as the foundation, sequencing data from 21 bloom Camellia, 24 oilseed Camellia, 41 wild Camellia and 15 other Camellia groups were extracted from the teabase database (Supplemental Table S3), followed by population structure analysis and principal component analysis. K values ranging from 1 to 9 and their corresponding cross-validation (CV) error values were examined (Fig. 3a; Supplemental Fig. S4). Notably, at K = 2, Hainan tea diverged from other Camellia species, manifesting distinct ancestral compositions. With K = 3, a subsequent separation occurred with Sinensis exhibiting novel ancestral components. By K = 4, Assamica segregated from the genetic makeup of the added Camellia plant samples. At this juncture, Hainan tea demonstrated a distinctive population genetic background compared to the global Assamica and Sinensis, corroborating the findings depicted in Fig. 2. Furthermore, at K = 8, the cross-validation error minimized (Fig. 3a), indicating the optimal model where multiple species within Hainan tea and Camellia were delineated into eight genetically discrete populations. Within these populations, the LMS group displayed an autonomous genetic composition (depicted in purple). Particularly noteworthy, populations from the LD-JFL and QZ-BML, originating from two rainforest reserves, exhibited a considerable blend of purple genetic backgrounds. Additionally, the NS-JM and SM-SM populations shared a common genetic makeup (depicted in dark brown). It’s of significance to observe that certain SM-MN populations encompassed a genetic background akin to global Assamica1, plausibly due to the historical introduction of Assamica in Hainan Province.
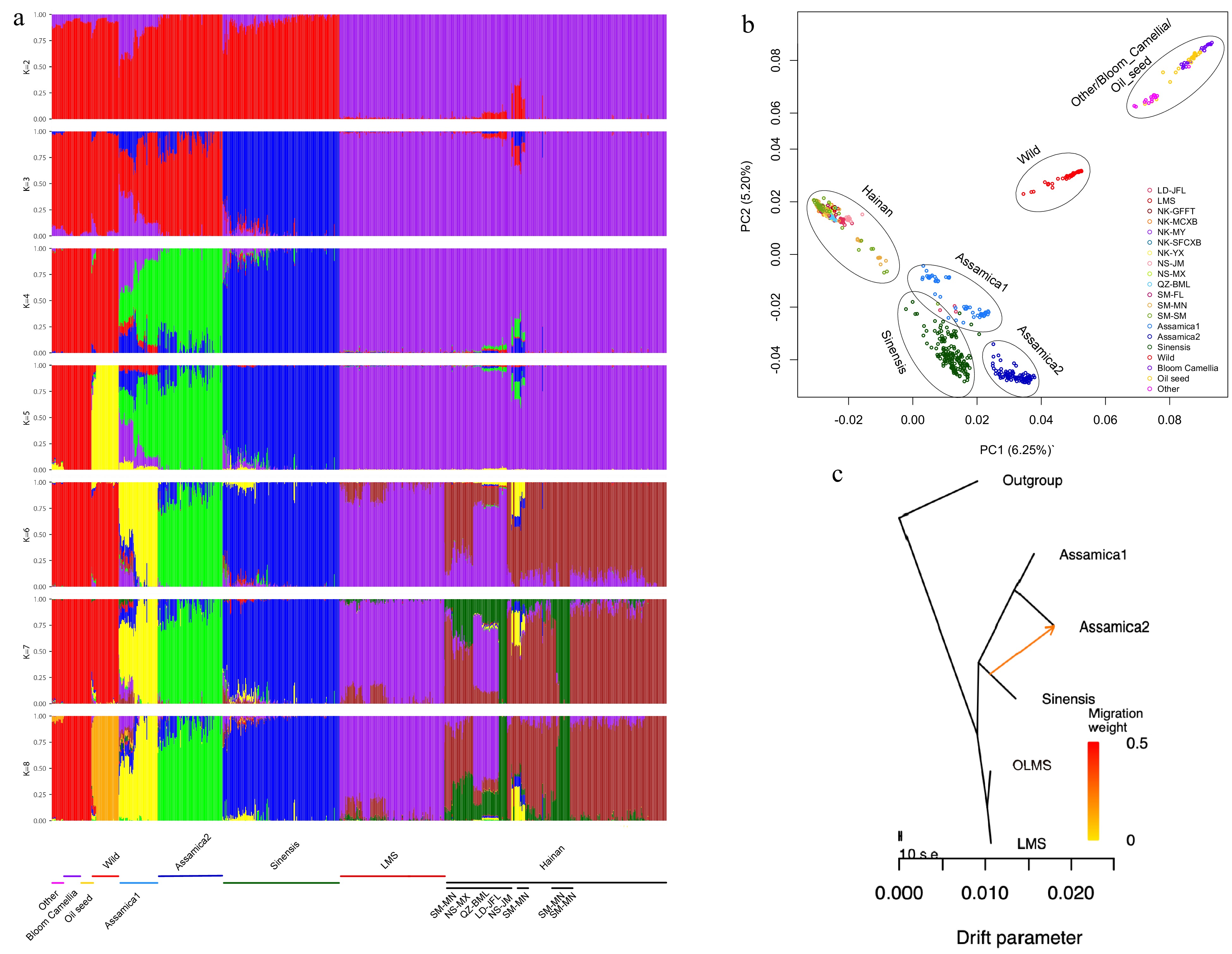
Figure 3.
The population structure and principal component analysis results for various species of Hainan tea and the Camellia genus are presented, alongside gene flow maps illustrating interactions between cultivated tea and Hainan tea.
(a) Population structure of Hainan tea and global Assamica, global Sinensis, 21 bloom Camellia, 24 oilseed Camellia, 41 wild Camellia, and 15 other Camellia groups. The picture shows the population structure of K value from 2 to 8 analyses. (b) Principal component analysis. These samples can be divided into six different groups, represented by six circles. Among them, Other, Bloom Camellia and Oilseed Camellia are grouped together, while Hainan Tea, global Assamica1, global Assamica2, global Sinensis, and wild Camellia are grouped separately. (c) Graph illustrating the results of the optimal gene flow analysis conducted using Treemix. This figure shows the history of gene flow among three populations of Assamica, Sinensis and Hainan tea. Arrows are used to mark possible population migration events in the figure, the length of the arrow indicates the intensity of migration, and the direction of the arrow indicates the direction of migration.In the principal component analysis of Hainan tea and Camellia species (Fig. 3b; Supplemental Fig. S5), PC1, PC2, and PC3 carried weights of 6.25%, 5.20%, and 4.53% respectively. PC1 distinctly segregated Hainan tea from other Camellia species, suggesting a unique genetic background for Hainan tea. This finding aligned with the outcomes of the population structure analysis (K = 2). PC2 separated global Assamica1 from Assamica2, while PC3 separated global Assamica from Sinensis. Consistent with the population structure results, PCA also indicated the inclusion of SM-SM samples within global Assamica1, further supporting the hypothesis that certain samples clustered within global Assamica was due to the introduction of CSA from Yunnan Province to Hainan Province. It’s noteworthy that Hainan tea displayed a closer clustering with Assamica and Sinensis in the PCA plot compared to the subsequently added Camellia genus. The findings from population structure and PCA complemented those of Fig. 2, demonstrating that Hainan tea not only stands distinct from Assamica and Sinensis but also lacks any shared genetic components with the newly added Camellia genus population.
Analysis of gene flow among Hainan tea, Assamica, and Sinensis
-
To provide additional evidence for our hypothesis proposing that Hainan tea belongs to a distinct species within the Camellia genus, separate from Assamica and Sinensis, we conducted f3 statistical analysis and examined the genetic relationships among Hainan tea, Assamica, and Sinensis using Treemix and D statistics.
During the f3 statistical analysis, we observed that the f3 values between Hainan tea and cultivated tea were highly similar, with a difference of only 0.00347, indicating a close genetic relationship between them. Specifically, the f3 values of OLMS and LMS exceeded those of Hainan tea and cultivated tea, implying a higher genetic similarity between OLMS and LMS. This discovery enhanced our understanding of the potential genetic connection between Hainan tea and LMS, suggesting a potential sharing of deeper genetic characteristics.
In the gene flow analysis, OptM determined the optimal migration model to be m = 1, implying the possibility of a migration event among the studied populations (Supplemental Fig. S6a, S6b). Employing a model with m = 1, we identified gene flow from Sinensis to Assamica1 (Fig. 3c), supported by a significant D value in the D statistics (Supplemental Table S5). In summary, these findings indicated that Hainan tea exhibits limited similarity to Assamica and Sinensis, with no significant historical gene flow between Hainan tea and Assamica or Sinensis.
Kinship analysis
-
In order to assess the genetic relationship between Hainan tea samples, KING software was used to analyze the genetic relationship of Hainan tea, Assamica and Sinensis, and the results are shown in Fig. 4a. In the genetic process, individuals accumulate their mutations, which can be shared among different individuals, so two individuals with the same mutations do not necessarily have the same ancestors. This similarity of mutations is called Identical by state (IBS). The King software can calculate the IBS value based on the SNPs, and reflect the composition and reliability of the kinship relationship among groups through the ratio of the kinship coefficient. The abscissa in the figure represented the proportion when IBS is 0, closer to 0, and higher the reliability of the result. The kinship coefficient of the vertical axis was mainly divided into three levels. A coefficient lower than 0.0442 indicated that the kinship relationship among individuals was far away, while a negative kinship coefficient indicated that there may be a large difference in the population structure between two individuals.
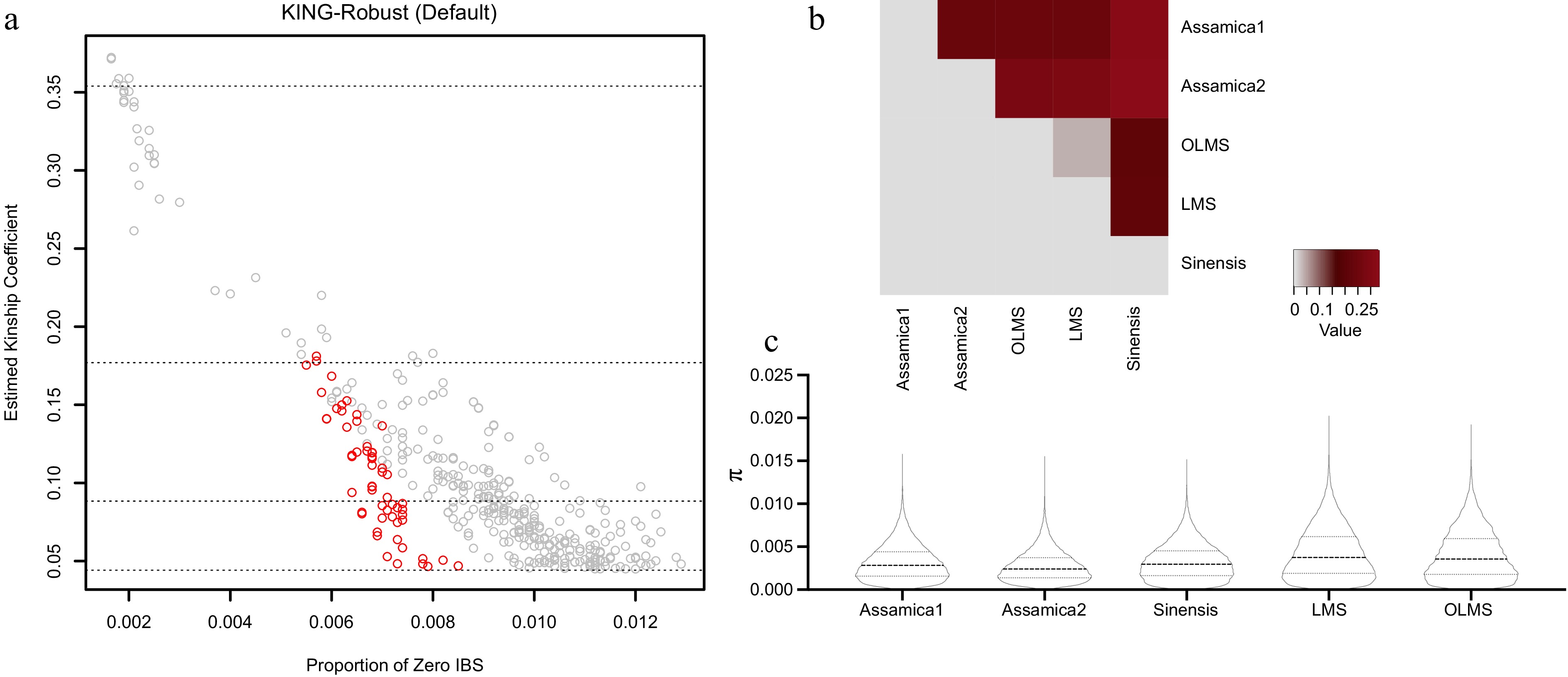
Figure 4.
The genetic relationships between samples and the genetic differentiation coefficient (FST) between different populations, as well as the corresponding nucleotide diversity (π).
(a) The family analysis results are based on KING software. The X-axis is the IBS value, and the Y-axis is the kinship coefficient. The data comprise pairs of samples exhibiting close kinship, which were selected from Global Assamica, Global Sinensis, and Hainan tea. Sample pairs from Hainan tea are highlighted in red. The three intervals, separated by dotted lines from top to bottom, represent primary, secondary, and tertiary degrees of kinship, respectively. (b) The FST heat map llustraties the genetic differences among the five populations of OLMS, LMS, Assamica1, Assamica2 and Sinensis. (c) π values of OLMS, LMS, Assamica and Sinensis populations.Highlighted in red in Fig. 4a was the sample pair with close genetic relationship screened out of Hainan tea samples, that was, kinship greater than 0.0442 (Supplemental Table S6). The closely related samples primarily belonged to the WDB group in the SM-MN area (refer to Fig. 4a; Supplemental Table S6 for specifics). All Hainan tea samples in this region were sourced from artificially managed tea gardens, with human activities influencing the reproduction of tea trees. Moreover, the area has a history of large-scale tea plant cultivation, suggesting that the close relationship between these samples may stem from the expansion of tea tree cultivation areas.
Analysis of genetic diversity of Hainan tea
-
Pairwise FST values were computed for five populations: Assamica1, Assamica2, Sinensis, Hainan tea, and LMS, revealing a range between 0.036 and 0.328 (Fig. 4b; Table 3). Notably, the minimal group distinction was observed between Hainan tea and LMS, resulting in an FST value of 0.036. Importantly, when compared to Assamica, the distinction between tea and Sinensis populations in the Hainan and LMS regions was less pronounced. This observation aligned with the findings from the phylogenetic analysis of f3. Genetic diversity levels for these five populations were additionally assessed through the analysis of π values. As illustrated in Fig. 4c, tea populations in the Hainan and LMS regions exhibited greater genetic diversity compared to Assamica and Sinensis. It is notable that teas from Hainan and LMS regions display heightened genetic diversity levels. The level of genetic diversity among tea populations is relatively consistent.
Table 3. Genetic differentiation coefficient (FST) among groups. FST values range from 0 and 1, with higher values indicating greater genetic differences among populations.
FST Assamica1 Assamica2 OLMS LMS Sinensis Assamica1 0.236 0.239 0.236 0.321 Assamica2 0.281 0.282 0.328 OLMS 0.036 0.209 LMS 0.212 Sinensis -
Although Hainan Island is rich in wild tea tree resources and possesses vast plantation areas of rainforest tea trees, tea tree resources have not yet been comprehensively investigated and fully developed. In this study, we selected a large number of ancient tea tree samples from the rainforest area, and analyzed them by whole genome resequencing, obtaining 32,334,340 SNPs. This dataset is the most extensive resequencing dataset of Hainan tea samples reported so far.
The classification of Camellia species basing on traditional taxonomy is very challenging[8], and Hainan dayezhong, as a unique Camellia species in Hainan, lacks the support of genomics data so far, and its status in taxonomy is always unknown so that it is often not yet a CSA. We analyzed the population relationship between Hainan tea and globally cultivated tea trees based on resequencing data to clarify the status of Hainan tea in Camellia from a genomic perspective. By constructing a phylogenetic tree between Hainan tea and globally cultivated tea trees, it can be observed that Hainan tea does not belong to either CSS or CSA, but rather forms an independent branch and clusters into a single taxon. It is important to note that in this cluster of Hainan tea, the samples from the LMS group formed distinct geographic subgroups, whereas the samples from the OLMS group did not appear to be geographically clustered (Fig. 2). This may be attributed to the fact that samples from the LMS population were collected in the Limu Mountain Rainforest Reserve, which is relatively undisturbed by human activities. In contrast, other areas have more human activities, which may lead to the mixing of genetic backgrounds of Hainan tea in multiple regions[16].
Although the Wuzhishan region is located in a tropical rainforest reserve, according to the Qiongzhong County Record, the state actively promoted tea planting in the region in the mid-1990s and introduced CSA varieties for breeding and cultivation. Therefore, the samples from the Wuzhishan region did not show obvious geographical clustering (Fig. 2). Additional results of population structure and principal component analysis further confirmed this observation. The population structure analysis revealed that Hainan tea has an independent genetic background, whereas LMS differs from OLMS in genetic background. It is particularly noteworthy that, except for LMS, OLMS presented a mixture of genetic backgrounds, which coincided with the results of phylogenetic trees (Figs 2, 3a). The results of principal component analysis also clearly showed the independent group status of Hainan tea with CSS and CSA (Fig. 3b; Supplemental Fig. S5). Despite the presence of several Hainan tea samples in the global Assamica1 cluster, this is consistent with the historical context of the introduction of CSA from Yunnan in the mid-1990s.
Geographic isolation is one of the main causes of species formation[17]. When populations of the same breeding stock separate, they face independent evolutionary histories defined by natural selection, genetic drift, adaptation, and colonization to local conditions[18]. Hainan, as a tropical island, has extensive rainforests that provide high-quality growing environments for plants, and the island’s geography provides the necessary geographic isolation for new species to arise. The results of the population structure analysis, which incorporated data from additional Camellia plants, clearly indicated that Hainan tea possesses a distinct genetic background compared to other Camellia species. Moreover, Hainan tea clustered closer to CSS and CSA in the principal component analysis while remaining distant from other Camellia (Fig. 3a, b). Therefore, we cautiously proposed that Hainan tea represents a novel variety of Camellia sinensis distinct from CSS and CSA. Notably, samples from the LMS region form a distinct subgroup cluster in the phylogenetic tree depicted in Fig. 2 and demonstrated an independent genetic component in the population structure analysis, akin to the scenario observed with G. hirsutum L. purpurascens on Hainan Island[19]. Thus, it was deduced that Hainan tea from the LMS region constitutes a unique endemic variety within the Hainan tea species.
Genetic drift is one of the important mechanisms for maintaining genetic diversity among biological populations. High levels of genetic drift help to reduce genetic differences and increase the homogeneity between two populations[20]. However, when physical barriers prevent genetic drift, different populations may form or experience physical isolation that prevents the exchange of genetic materials. These physical barriers are usually, although not always, caused by natural factors[21].
Hainan Island, once connected to the mainland, has undergone a long period of rotation and movement, rotating counter-clockwise from its original position in the Beibu Gulf to its current position. The initial separation occurred in the Paleocene (ca. 65 Mya), while the major part of the rotational drift occurred in the Eocene[22]. During the Quaternary, ice ages and interglacial periods alternated, the most recent major ice age occurring about 15 Kya ago. The onset of the Ice Age led to a drop in global temperatures and a steady decline in global sea levels, which led to the formation of natural land bridges between sea islands and continents. During the ~8,000-year-long Ice Age (15 Kya-7 Kya ago), genetic exchange of species between Hainan Island and neighboring continents may have occurred. For example, a literature survey study found the existence of gene flow between Hainan’s native Painted Lady and the Chinese Painted Lady in South China[23]. However, the cold global climate during the Ice Age reduced the population size of the species, especially for the cold-intolerant CSA, and the likelihood of genetic exchange diminished[24]. After the Ice Age ended, the rise of the sea level led to the emergence of Qiongzhong Strait, which switched Hainan Island once again to the island mode. This geological event may have hindered genetic exchange between Hainan Island tea trees and those on the mainland, leading to their gradual and independent evolution in response to the tropical island climate. As a result, a new variant emerged, possibly falling under the categorization of Camellia sinensis[25].
Considering the potential existence of land bridges facilitating gene flow between Hainan tea and mainland tea plants, we intensively investigated the gene flow between Hainan tea and cultivated tea. First, we performed f3 statistical analysis (Supplemental Table S4) and found that the genetic relationship between LMS and OLMS was closer comparing to cultivated tea, which is consistent with the results in Fig. 2 and 3a. Especially noteworthy is that Hainan tea was closer to Sinensis comparing to Assamica. The results of the Treemix analysis visually demonstrated how geographic isolation significantly impeded gene flow between cultivated and Hainan teas (Fig. 3c). In addition, the Dsuite program was applied to perform ABBA-BABA analysis, and this result further supported our view (Supplemental Table S5). These findings strongly suggested that the geographic separation of Hainan tea has prevented the exchange of genetic material between it and cultivated tea, thus contributing to its possible independent evolution as a new variant of Camellia sinensis.
Groups that are highly segregated and lack genetic drift are usually prone to inbreeding[26]. However, the current analyses showed (Fig. 4a) that Hainan tea do not show excessive kinship among each other, and the concentration of samples with high kinship was overwhelmingly from samples from the WDB group (Supplemental Table S6), a group whose tea trees came from an artificially managed tea plantation. This phenomenon may be caused by anthropogenic factors. Genetic diversity, species diversity, and ecosystem diversity are the three pillars of biodiversity. Tea plants are typically propagated asexually via cuttings. If individuals propagated through this method are presented in the study samples, a significant portion of sample pairs will exhibit an affinity coefficient exceeding 0.354. Nevertheless, the present findings do not corroborate this hypothesis (Fig. 4a). Based on the principles of population genetics, the conservation of biodiversity is ultimately the conservation of genetic diversity[27]. Nucleotide diversity is an important indicator for assessing the diversity of DNA sequences in a species or population[28]. The processes of domestication and breeding have reduced the genetic diversity of crops, and the widespread cultivation of monoculture crop varieties has led to an increase in genetic vulnerability[29,30]. Wild ancient tea trees, as a precious natural resource with high genetic diversity, are of great value for the study of the evolutionary mechanisms and diversity of the tea trees[31]. Interestingly, the Hainan tea and LMS have higher genetic diversity than CSS and CSA (Fig. 4c), even though Hainan tea is affected by geographic isolation, resulting in restricted gene flow (Fig. 3c). This can be partially attributed to the unique climatic conditions of the tropical island, which are very favorable for the growth of tea trees. Combined with minimal anthropogenic disturbance, this has resulted in less natural pressure on tea tree population expansion, thus helping to maintain genetic diversity[16]. Furthermore, the genetic relationships between tea plants in Hainan and LMS were closer to those of Sinensis than to Assamica, and the genetic relationships between tea trees in Hainan and LMS were closer to each other (Fig. 4b; Table 3). This is consistent with the results obtained in the f3 statistical analysis (Supplemental Table S4), suggesting that Hainan tea and Assamica taxa diverged earlier than Hainan tea and Sinensis taxa.
In summary, the whole-genome resequencing of 500 Hainan tea samples from major tea-producing regions of Hainan Island was performed in this study, and 32,334,340 SNPs were successfully identified. The results of this study strongly support the existence of Hainan tea as a new variant of Camellia sinensis, which is genetically distinct from CSS and CSA, and also reveal the existence of Hainan tea in the LMS region as an independently evolved local variety. Although Hainan tea did not show significant gene flow between Hainan tea and cultivated tea trees due to the geographic barrier of the strait, it still maintained high genetic diversity, which manifested itself in high π values. The results of this study help to clarify the position of Hainan tea in the taxonomy of Camellia sinensis from a genomic perspective. Additionally, they provide reliable data support for an in-depth understanding of the genetic background and diversity of Hainan tea on the island. Furthermore, they offer an important scientific basis for the conservation of tea germplasm resources and molecular breeding on Hainan Island. In addition, our research methods and techniques can also provide lessons and references for the analyses of the origin and domestication of other species on Hainan Island, as well as for genetic diversity studies.
-
Systematically, 500 samples of Hainan tea from Hainan Province, China were collected. These included Jianfengling (Ledong, 28 samples), Limu Mountain (Qiongzhong, 160 samples), Gaofeng Fangtong Village (Nankai, Baisha, 41 samples), Miao Village Junior Class (Nankai, Baisha, 53 samples), Mengya Village (Nankai, Baisha, 24 samples), Shifu Village Junior Class (Nankai, Baisha, 26 samples), Yaxing (Nankai, Baisha, 15 samples), Junmin Village (Nansheng, Wuzhishan, 13 samples), Maoxiang Village (Nansheng, Wuzhishan, 32 samples), Baimaling (Qiongzhong, 13 samples), Fanglong Village (Shuiman, Wuzhishan, 14 samples), Maona Village (Shuiman, Wuzhishan, 47 samples), and Shuiman Village (Shuiman, Wuzhishan, 34 samples). Additionally, the teabase database[15] and data from various Camellia species in the genome sequence archive with project number PRJCA001158 from Genome Sequence Archive[8] were utilized for analysis. Notably, the KM6 strain (Cuspidata Camellia) was selected as an outgroup for our study and subsequent analyses. Detailed information on the study samples can be found in Supplemental Tables S1, S3, and Fig. 1.
DNA sample preparation and sequencing
-
Five hundred tea accessions were acquired exclusively from Hainan province in China. Young leaves were harvested from these plants and rapidly frozen in liquid nitrogen. Total DNA extraction was performed using the DNAsecure plant kit (Tiangen, Beijing). Subsequently, 2 µg of genomic DNA from each accession was utilized to prepare sequencing libraries according to the manufacturer’s protocol using the NEBNext Ultra DNA Library Prep Kit (NEB Inc., America). Sequencing was carried out on an Illumina NovaSeq 6000 sequencer, generating paired-end sequencing libraries with an approximate insert size of 400 bp.
Quality control and filtering
-
The paired-end resequencing reads underwent filtering utilizing fastp (Version: 0.12.2)[32]. This process eliminated reads containing adapter sequences or poly-N sequences, as well as low-quality reads (defined as reads with more than 40% bases having Phred quality scores ≤ 20) from the raw data. The outcome of this step was the production of clean data, which were then utilized for subsequent downstream analyses.
Variation calling and annotation
-
The paired-end resequencing reads were aligned to our tea reference genome using BWA (Version: 0.7.17-r1188)[33], employing default parameters. The mapping results were converted into the BAM format and unmapped as well as non-unique reads were filtered using SAMtools (Version: 1.3.1)[34]. Additionally, duplicated reads were removed using the Picard package (picard.sourceforge.net, Version: 2.1.1).
Following BWA alignment, we performed realignment of reads around indels using GATK in a two-step process. Initially, the RealignerTargetCreator package was utilized to identify regions necessitating realignment. Subsequently, the identified regions were realigned using IndelRealigner, resulting in a realigned BAM file for each accession. Variant detection was conducted following the recommended best practice workflow by GATK[35]. Specifically, variants were called for each accession using the GATK HaplotypeCaller[35]. A joint genotyping step was carried out to merge variations comprehensively from the gVCF files. During the filtering step, the SNP filter expression was set as ‘QD < 2.0 || MQ < 40.0 || FS > 60.0 || SOR > 5.0 || MQRankSum < −12.5 || ReadPosRankSum < −8.0 || QUAL < 30’. SNPs that were not bi-allelic were excluded, resulting in the creation of the basic set. Subsequently, SNPs with more than 20% missing calls and MAF less than 0.05 were further eliminated to generate the core set, which was used for phylogenetic tree construction, PCA, and population structure analysis.
SNPs were annotated according to the tea genome using the ANNOVAR package (Version: 2015-12-14)[36]. Based on the genome annotation, SNPs were classified into various genomic regions, including exonic regions (overlapping with coding exons), splicing sites (within 2 bp of a splicing junction), 5' UTRs, 3' UTRs, intronic regions (overlapping with introns), upstream and downstream regions (within a 1 kb region upstream or downstream from the transcription start site), and intergenic regions. SNPs located in coding exons were further categorized into synonymous SNPs (which did not cause amino acid changes), nonsynonymous SNPs (which caused amino acid changes), stop gain mutations (mutations resulting in the gain of a stop codon), and stop-loss mutations (mutations resulting in the loss of a stop codon). Indels within exonic regions were classified based on whether they caused frame-shift mutations (3 bp insertion or deletion) and whether they resulted in the gain or loss of a stop codon.
Population genetics analysis
-
Whole-genome SNPs were utilized to construct the maximum likelihood (ML) phylogenetic tree with 100 bootstrap replicates using SNPhylo (Version: 20140701)[37]. Camellia cuspidata (KM6) served as an outgroup to provide corresponding positional information. The phylogenetic tree was visualized and color-coded using iTOL (
http://itol.embl.de ).Chromosomal SNPs were filtered by removing SNPs in linkage disequilibrium with PLINK (Version v1.90b3.38)[38] , employing a window size of 50 SNPs (advancing 1 SNP at a time) and an r2 threshold of 0.5. Principal component analysis was conducted using Genome-wide Complex Trait Analysis (GCTA, version: 1.25.3) software[39] , and the first three eigenvectors were plotted. Population structure analysis was performed using the ADMIXTURE program (Version: 1.3)[40] with a block-relaxation algorithm. The number of genetic clusters (K) was predefined from 2 to 9, and the cross-validation error (CV) procedure was run to explore convergence of individuals. Default methods and settings were applied in all analyses.
Relationship inference
-
The relationship between each accession was examined using KING (Version: 2.2.5)[41] , utilizing the basic set SNPs with the option ‘--kinship’. This option employed the KING-Robust algorithm to estimate pair-wise kinship coefficients. Close relatives were reliably inferred based on the estimated kinship coefficients using the following simple algorithm: an estimated kinship coefficient range greater than 0.354 indicates a duplicate relationship, while ranges of [0.177, 0.354], [0.0884, 0.177], and [0.0442, 0.0884] correspond to 1st-degree, 2nd-degree, and 3rd-degree relationships, respectively.
Genetic variation and FST calculations
-
The calculation of average pairwise diversity within each population (π) was conducted using 100 kb sliding windows. Population differentiation (FST) was assessed through pairwise FST comparisons among populations.
Gene flow analysis
-
Admixture graphs of geographically defined Hainan tea populations were inferred using TreeMix[42], employing a Maximum Likelihood (ML) approach based on a Gaussian model of allele frequency change. The topology of the ML trees varies depending on the number of migration events (m) permitted in the model, ranging from m = 0 to m = 5. Bootstrap values on the tree were derived from 1,000 replicates. Admixture events among different tea populations were indicated by arrows on the graph, with KM6 serving as the root. To ensure robustness, each migration event was iterated 10 times with a random seed. The optimal number of migration edges was determined using the R package ‘OptM’ (Version: v0.1.6)[43].
f3 and Patterson’s D statistics
-
The f3 statistics were computed using the R package ‘admixr’ (Version: 0.9.1)[44] for all conceivable combinations of tea groups, with KM6 serving as the outgroup. SNPs exhibiting missing data and monomorphism were excluded from the analysis.
To assess the presence of introgression signals among tea groups, Patterson’s D (also known as the ABBA-BABA test) and f4 admixture ratio statistics for all possible trios of tea groups were calculated using Dtrios in Dsuite (Version: 0.4 r42)[45], with KM6 designated as the outgroup. SNPs with missing data and monomorphism were removed from consideration.
To investigate the species-level relationships among tea groups, we explored the backbone of the phylogeny using the PoMo model[46] within IQ-Tree[47]. This analysis included 1,000 bootstrap replicates, employing the ultrafast bootstrap approximation method. The tree was rooted using KM6 as the outgroup.
-
The authors confirm contribution to the paper as follows: study conception and design: Duan S; draft manuscript preparation: Guo D, Li D; manuscript revision and editing: Huang Y, Duan S; tea samples collection: Wang Z, Li D, Zhou Y, Xiang G, Zhang W, Wang W, Fang Z, Hao T, Zheng D, Lei Y, Yang L, Zhang W, Tang S, Zheng L, Cao Y. All authors reviewed the results and approved the final version of the manuscript.
-
The data that supported the findings of this study have been deposited into the CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) with accession number CNP0005405. In addition, the sequencing data are also accessible from the tea database (
http://teabase.ynau.edu.cn/index/download/index ) and the BIG Data Center under the accession number PRJCA001158. This work was supported by the Hainan Academy of Agricultural Sciences Research Project (HAAS2022KJCX03), Research and Demonstration on Key Technologies of Germplasm Resource Bank Construction and Resource Innovation Utilization of Wuzhishan Big Leaf Tea (ZDYF2024XDNY245) and Monitoring and Analysis of Key Quality Components of Hainan Big Leaf Black Tea and Development and Demonstration of New Standardized Processing Technology (WZSKTPXM202202). We express our sincere gratitude to the People’s Government of Wuzhishan City, Hainan Province, and the Wuzhishan Scenic Area of Hainan Tropical Rainforest National Park for their generous support and assistance in this project.
-
The authors declare that they have no conflict of interest.
-
#Authors contributed equally: Dazhong Guo, Dongliang Li
- Supplemental Table S1 The relevant information of 500 Hainan tea samples used in this study.
- Supplemental Table S2 Quantity table of SNPs after different hard filter.
- Supplemental Table S3 Supplementary Table 3. The information used in this study includes global Assamonia, global Sinensis, Other, Bloom Camellia, Oil seed, and Wild samples.
- Supplemental Table S4 f3 Statistical analysis results.
- Supplemental Table S5 D Statistical analysis results.
- Supplemental Table S6 Hainan tea samples used in KING software analysis of genetic relationships.
- Supplemental Fig. S1 The density plot of the sample's mapping rate, X-coordinates indicate the Mapping rate and Y-coordinates indicate the density.
- Supplemental Fig. S2 Density distribution plot of SNPs hard filter.
- Supplemental Fig. S3 Group rootless trees constructed by OLMS, LMS, global Assamica and global Sinensis. KM6 (C. cuspidata) was selected as the outgroup.
- Supplemental Fig. S4 Figure 3A cross-validation error. The x-axis represents the K value, and the y-axis represents the cross-validation error. The dots in the figure show K = 8 with the smallest cross-validation error.
- Supplemental Fig. S5 Principal Component Analysis of Hainan Tea with global Assamica, global Sinensis, wild Camellia sinensis, Bloom Camellia, Oil seed Camellia and other Camellia. Principal Components 1 and 3. The analysis focused on principal components 1 and 3. Each sample is represented by a small circle, with the color of each circle indicating the corresponding group as depicted in the legend on the right side.
- Supplemental Fig. S6 (A,B) Optimal m-value diagrams for OptM judgment. The horizontal axis is the m value, and the vertical axis is the likelihood value. The optimal m value in the figure is 1, which means that the historical relationship of samples can be best explained by 1 migration.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Yunnan Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Guo D, Li D, Wang Z, Li D, Zhou Y, et al. 2024. Genome resequencing reveals an independently originated Camellia sinensis variety – Hainan tea. Agrobiodiversity 1(1): 3−12 doi: 10.48130/abd-0024-0003 

Genome resequencing reveals an independently originated Camellia sinensis variety – Hainan tea
- Received: 04 March 2024
- Revised: 27 March 2024
- Accepted: 11 April 2024
- Published online: 17 May 2024
Abstract: Tea, originating in China over 3,000 years ago, has transitioned from a medicinal herb to a widely consumed beverage. Despite considerable research focusing on tea plants in southwestern China, little attention has been paid to those on Hainan Island. The notable resemblance between Hainan tea and C. sinensis var. assamica, alongside the unique geographical and climatic conditions of Hainan Island, has presented significant challenges for taxonomic and genetic investigations concerning Hainan tea. Our study bridged this gap by collecting 500 samples from Hainan Province and employing whole-genome resequencing to examine interspecific differences between Hainan tea and cultivated varieties. The findings confirmed the distinct taxonomic position of Hainan tea within Camellia sinensis, providing valuable insights for resource conservation and molecular breeding. Furthermore, our methodology offers a framework for investigating the origin, domestication, and genetic diversity of other species native to Hainan Island.
-
Key words:
- Camellia sinensis /
- SNPs /
- Whole genome resequencing /
- Population structure /
- Hainan