









-
Cultivated for millennia, grapevines (2n = 2x = 38) rank among the earliest documented examples of plant domestication, dating back approximately 22,000 years[1]. Grapevines play a crucial role not only in human health but also in maintaining ecological diversity. Covering over 75,866 square kilometers globally (Food and Agriculture Organization of the United Nations, 2021), grapevines stand as one of the world's four major fruit tree crops. Driven by this importance, advancements in sequencing technology have spurred the de novo assembly and analysis of numerous grape genomes, including those of cultivated varieties like 'Cabernet Sauvignon'[2], 'Cabernet Franc'[3], 'Carmenere'[4], 'Chardonnay' , 'Shine Muscat'[5], as well as wild grape species such as V. riparis[6], V. rotundifolia[7], V. amurensis[8], and V. adenoclada[9]. By elucidating these genomic landscapes, researchers aim to propel both hybrid breeding and population genetics research.
Leveraging genome assembly, whole genome sequencing (WGS) and population genomics, significant progress has been made in understanding the grapevine genetic characteristics. For instance, a study analyzing 472 individuals identified selection signals linked to population demographic history, fruit edibility, and stress resistance, while also pinpointing candidate genes associated with fruit shape and other traits[10]. Another study explored the functional impact of chromosomal structural variation, demonstrating how an inversion on chromosome 2 influences grape sex and skin color, highlighting the importance of such variations in domestication[11]. Furthermore, analysis of 3,304 WGS data provided insights into two distinct grape domestication events, characterizing both genetic diversity and domestication signatures[12]. However, despite these advancements, research has primarily focused on understanding grape traits and their domestication history, with limited exploration of population genomic studies in grape hybrid breeding.
'Shine Muscat', a popular interspecific hybrid grape (V. labrusca × V. vinifera) originating from Japan, has gained widespread cultivation, particularly in China[13]. This table grape cultivar is renowned for its large, yellow-green berries with crisp flesh, distinct muscat flavor, high sugar content, and low acidity[5]. In contrast, V. adenoclada Hand. Mazz., a wild grape species native to China thrives in the regions south of the Yangtze River[9,14]. This species exhibits remarkable resilience to high temperatures, humidity, fungal diseases, drought, and poor soil conditions[15]. It is also rich in phenolic acids and flavanols, and has played a key role in Guangxi Province's grape industry by enabling the cultivation of excellent varieties and establishing a thriving winemaking tradition in China[9]. The desirable phenotypic traits of both 'Shine Muscat' and V. adenoclada make them valuable parental candidates for further breeding endeavors.
Hybridization is of great significance for crop breeding, especially in the current era of smart crop breeding 4.0[16]. Grape hybridization allows for the development of new grape varieties with improved characteristics such as disease resistance, yield, flavor, and adaptability to different climates and soil conditions. Hybridization between 'Shine Muscat' and V. adenoclada may play a crucial role in grape breeding by facilitating the integration of favorable traits and increasing genetic diversity. Currently, it is widely suggested that numerous generations of domestication and breeding might cause the alteration of deleterious mutations, which have the potential to affect the general fitness of organisms[17,18]. Various ways of propagation during breeding, such as crossing, selfing, or even apomicts, will have different effects for deleterious load accumulation[19]. Therefore, it is interesting to figure out what hybridization could do to the genetic load of offspring.
In this study, 28 accessions were obtained that consist of cultivated 'Shine Muscat' varieties, wild species V. adenoclada, and their one progeny. We aimed to explore the specific characteristics among these accessions, including the genetic structure, genetic differentiation, selected loci, and genetic burden. The present study will serve as a foundation for the sustainable development of the grape industry and the improvement of grape varieties.
-
In total, 28 whole genome sequencing (WGS) individuals were used in this study. There are 13 samples originated from a common garden in Guangxi Province, China, comprising 11 wild V. adenoclada accessions (ADE1-ADE11; collected from a wild population in Nanning, Guangxi, China), two V. labrusca × V. vinifera 'Shine Muscat' accession (MUS1, MUS7), and one hybrid offspring accession (HYB1) through hybridizing V. adenoclada (ADE1) and 'Shine Muscat' (MUS1). Besides, nine wild V. adenoclada accessions (ADE12-ADE20) were collected in Guangdong Province, China. These samples were all newly sequenced in this study. Besides, five 'Shine Muscat' accessions (MUS2−MUS6)[5] data were downloaded from the National Center for Biotechnology Information (NCBI). All V. adenoclada accessions were clustered into the ADE group below, and all 'Shine Muscat' accessions were regarded as members of the MUS group. This diverse sample composition enables investigation and potential elucidation of the genetic relationships between these two Vitis species and their hybrid offspring.
Sequencing and variants calling
-
The genomic DNA of all samples was isolated from leaf tissues using the Qiagen Dneasy plant kit. Adhering to the Illumina library construction protocol, paired-end sequence libraries were constructed with an insert size of 300−400 bp. Libraries were then sequenced using Illumina NovaSeq 6000 with 2 × 150 bp paired-end reads. The raw sequencing data newly generated in this study have been deposited in the NCBI with the BioProject ID of PRJNA1082482.
After sequencing, fastp (v0.23.2)[20] was utilized with default parameters to filter out the adapters and low-quality reads. Then, the trimmed high-quality reads were aligned to the PN_T2T reference genome[21] with BWA-MEM algorithm (v0.7.17-r1188)[22], with duplicated reads sorted and removed by SAMtools (v1.13)[23]. The 'vc' function implemented in GTX was used for single nucleotide polymorphism (SNP) and genotype calling for all 28 samples[22]. After gaining the gvcf file for each sample, the 'joint' function in GTX was used to join SNPs together. Finally, we used VCFtools (v0.1.16) to reduce false positives with parameters: '-minGQ 20 -min-alleles 2 --max-alleles 2 -maf 0.001 -max-missing 0.8'. These parameters entail removing SNPs with quality scores lower than 20, eliminating SNP sites containing more or fewer than two alleles, restricting the minimum allele frequency to 0.001, and discarding SNPs with over 20% missing genotypes across all samples.[24]
Population structure
-
To construct a phylogenetic tree, admixture analysis, and principal component analysis (PCA), PLINK (v1.90b6.21) with parameter '--indep-pairwise 50 10 0.5' further filter Linkage Disequilibrium (LD) SNPs was used[25]. The phylogenetic tree was conducted by FastTree (v2.1.10) with general time reversible models, and the resulting tree was visualized using the online tool iTOL (Interactive Tree of Life v3,
https://itol.embl.de )[26]. Individual admixture proportions were estimated using ADMIXTURE (v1.3,https://github.com/stevemussmann/admixturePipeline ) with K ranging from 2 to 4[27]. Principal component analysis (PCA) was performed by PLINK (v1.90b6.21) with parameter '--pca'. Identity-by-descent (IBD) was calculated among all 28 Vitis accessions using PLINK. To visualize the patterns of IBD sharing, networks were generated using Cytoscape (v3.6.0,www.cytoscape.org ).Selection analysis
-
Selscan (v1.2) with the default settings was used for calculating XP-EHH values of SNPs, which could specifically identify ongoing or nearly fixed selective sweeps by contrasting haplotypes between two populations[28]. The principle of XP-EHH (cross-population extended haplotype homozygosity) selection signal detection is based on the difficulty of breaking existing mutation segments due to linkage disequilibrium, while new mutations require a longer selection period to reach a high gene frequency. Therefore, when a certain haplotype segment appears at a high frequency in a population, it indicates that the gene segment has undergone selection and can be identified using the XP-EHH selection signal method. When calculating XP-EHH, 'Shine Muscat' population (MUS) was used as reference population. SNPs with XP-EHH scores greater than 2 and located in the top 1% were considered significantly selected in V. adenoclada (ADE), while those with scores less than −2 and in the bottom one percent were considered significantly selected in 'Shine Muscat'. Windows were defined as 20 kb, and the mean XP-EHH scores were calculated by the selscan norm function for each window.
Gene Ontology enrichment
-
Gene Ontology enrichment (GO) analyses were conducted using the DAVID online platform (
https://david.ncifcrf.gov/tools.jsp ). Initially, all protein sequences from the PN_T2T reference genome were aligned to the Swissport protein database (https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/swissprot.gz ) by diamond blastp[29] command, and the whole genome successfully blasted genes were considered background genes. Genes under selection were regarded as target genes, and submitted to the DAVID website for gene function annotation, allowing for the examination of gene function and enrichment pathway results.Population deleterious mutations
-
Candidate deleterious alleles were identified using SIFT4G (sorting intolerant from tolerated)[30]. Before annotation, we phased our SNP dataset by Beagle (v5.4), then flipped it based on the ancestral populations Muscadinia running Model 1 in the superSFS (
https://github.com/xhchauvet/superSFS ) script with parameter 3. Model 1 means only speculate the ancestral allel and output new vcf file using speculated allel as reference. Used PN_T2T genome and annotation to make a SIFT database with SIFT (https://github.com/pauline-ng/SIFT4G_Create_Genomic_DB ) predictions, which were applied to annotate the SNP files. A nonsynonymous position with a SIFT score below 0.05 was deemed as potentially deleterious SNPs. -
Following variant detection and strict filtering processes, a collection of 22,072,042 high-quality single nucleotide polymorphisms (SNPs) was retained from the 28 Vitis accessions for subsequent analyses. The whole-genome resequencing data in the present study had an average depth of 47 ×. And there are 23 samples newly sequenced, which ranged from 17.52 gigabases (Gb) to 24.06 Gb. The base pair mapping rates for the ADE and MUS groups were consistently high, spanning from 80.59% to 98.92%, with the majority exceeding 96%. A higher number of SNPs were observed within the wild ADE population compared to the cultivated MUS counterparts. Notably, the HYB hybrid accession exhibited the highest SNP count, suggesting greater genetic variation and mixed genetic background.
Genetic structure of hybrid offspring and progenitor accessions
-
To better understand the relationship of the 28 samples from 'Shine Muscat', V. adenoclada, and their progeny, an unrooted maximum likelihood (ML) tree was constructed with 28 accessions (Fig. 1a). As is shown from the tree, the wild V. adenoclada individuals and cultivated 'Shine Muscat' varieties were well divided into two clades, with their hybrid progeny (HYB1) intermediate between them.
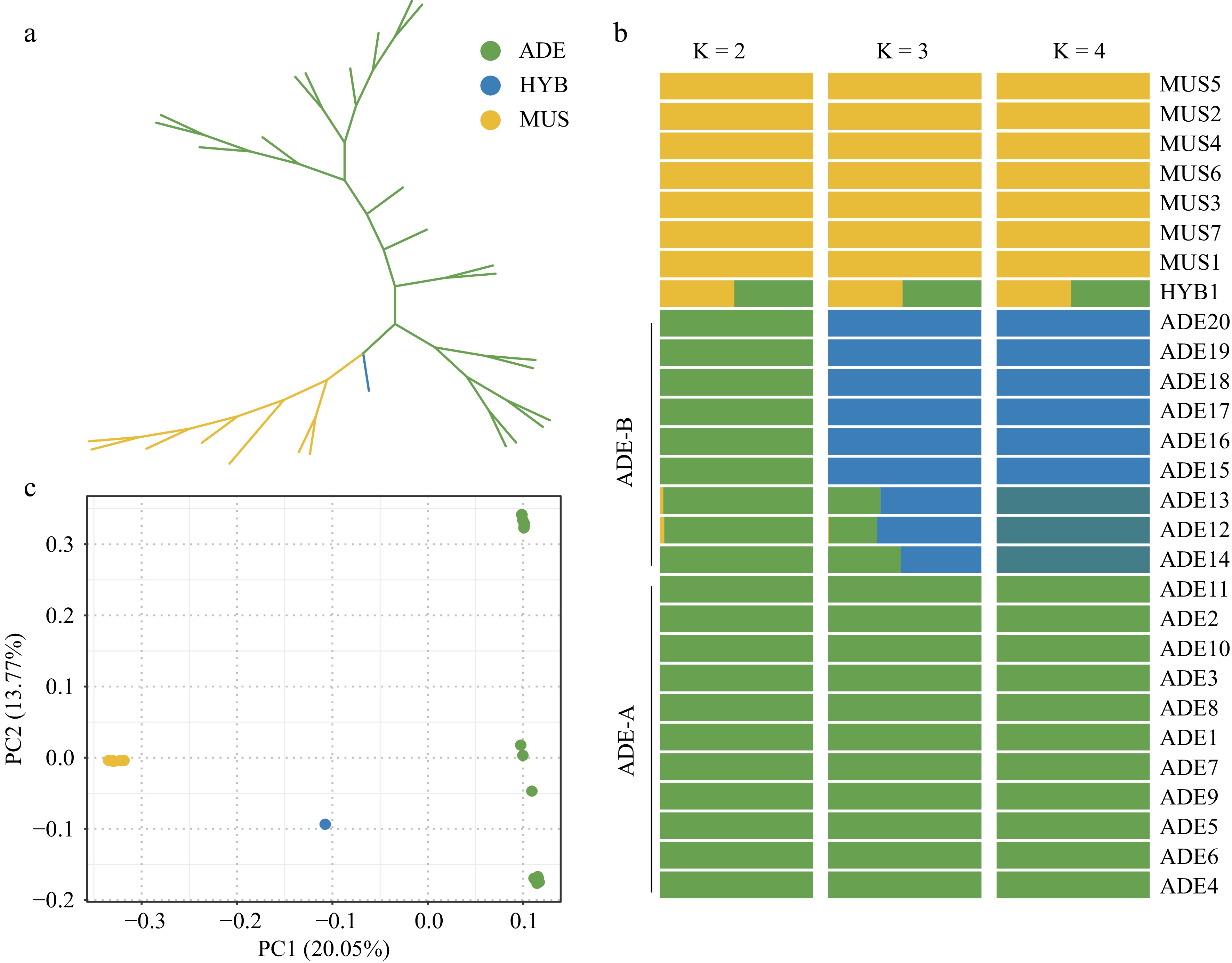
Figure 1.
Population structure of the 28 Vitis accessions. (a) Unrooted ML tree. The colors yellow, green, and blue represent the 'Shine Muscat' group, the V. adenoclada group, and the hybrid offspring, respectively. (b) Admixture analysis with K = 2, 3, and 4. (c) PCA results. The correspondence between colors and groups is consistent with Fig. 1a.
The ancestry source of these accessions was inferred through admixture (Fig. 1b) with K = 2, 3, 4. When K was equal to 2, the Vitis accessions could roughly be divided into two groups, which was represented respectively by MUS and ADE. It was apparent that HYB1 showed a sign of hybridization of two groups, which was consistent with the phylogenetic tree. When K was 3, the ADE group was further divided mainly into two parts, which distinguished individuals in different geographical distribution. Three ADE accessions in Guangdong Province (ADE_B) was further diverged as K = 4.
PCA results were concordant with the phylogeny and population structure analysis that MUS group, ADE group, and HYB1 were clustered into three independent parts (Fig. 1c). PC1 well divided these three parts. HYB1 was in the middle of ADE and MUS groups in the PCA plot, which indicated its specificity relative to parental accessions. ADE group was relatively scattered, which was also consistent with the admixture results when K = 3 and 4.
The relatedness among these accessions was further inferred using identity by descent (IBD; Fig. 2). Three IBD clusters were roughly identified from the network, namely the ADE group and the MUS group. IBD scores between ADE_A accessions (ADE1−ADE11) were generally higher than 0.7, which suggested that these accessions had a closer kinship, like parent-child or siblings (Supplemental Table S1). In comparison, the IBD score calculated within ADE_B accessions exhibited lower IBD scores (below 0.6). Because of the clonal reproduction property, MUS possessed exceedingly high IBD scores (higher than 0.9) within the group. HYB1 had a higher IBD score (0.5) with ADE1 than calculated with other ADE accessions, and meanwhile, HYB1 exhibited a relatively closer kinship with MUS1. Given the hybrid experiment conducted with ADE1, and MUS1, this result reflected the parent-child relationship between ADE1, MUS1, and HYB1.
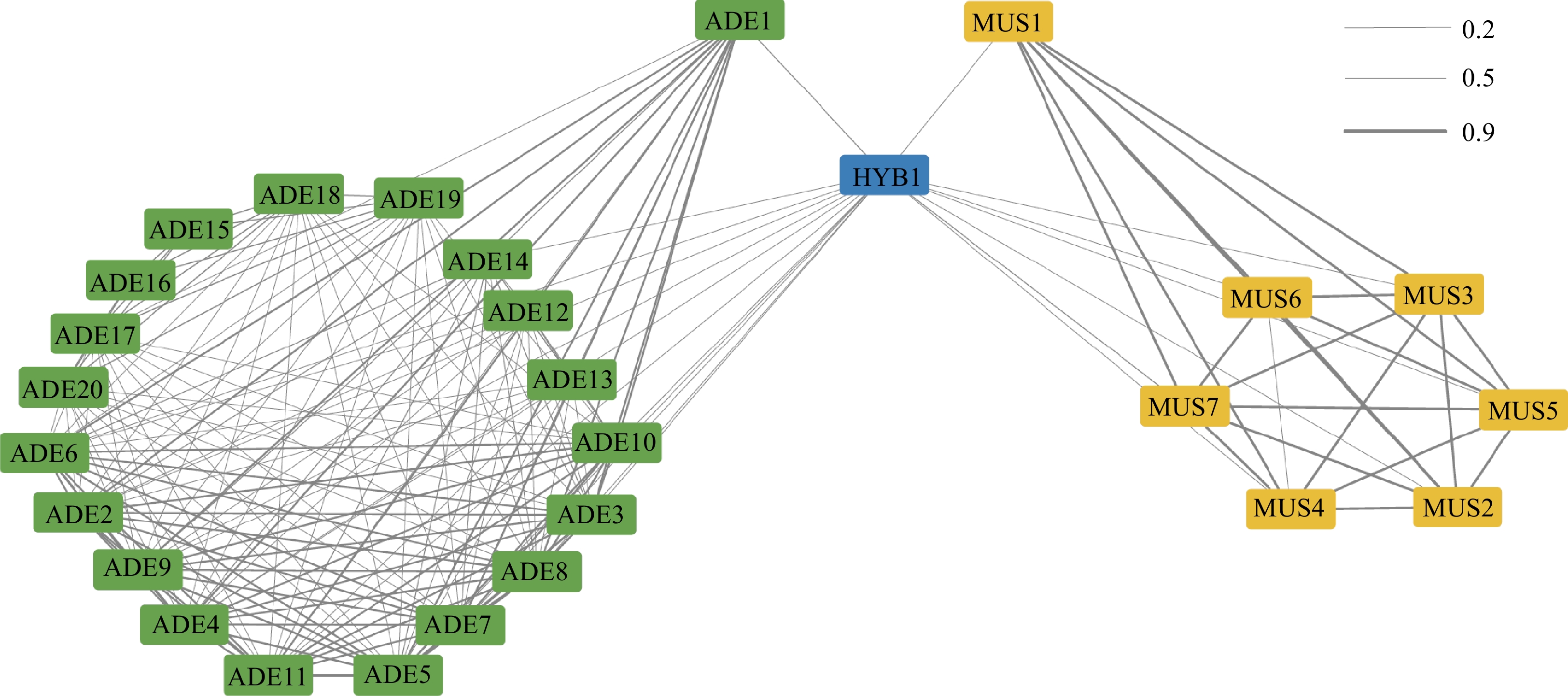
Figure 2.
IBD Relationship among the Vitis accessions.
Selection sweeps signals in V. adenoclada and Shine Muscat
-
XP-EHH analysis was conducted to investigate SNPs and regions that were under positive selection between V. adenoclada and 'Shine Muscat' (Fig. 3). Selected SNPs in V. adenoclada were distributed across 60 windows, whereas selected SNPs in 'Shine Muscat' were distributed across 362 windows.
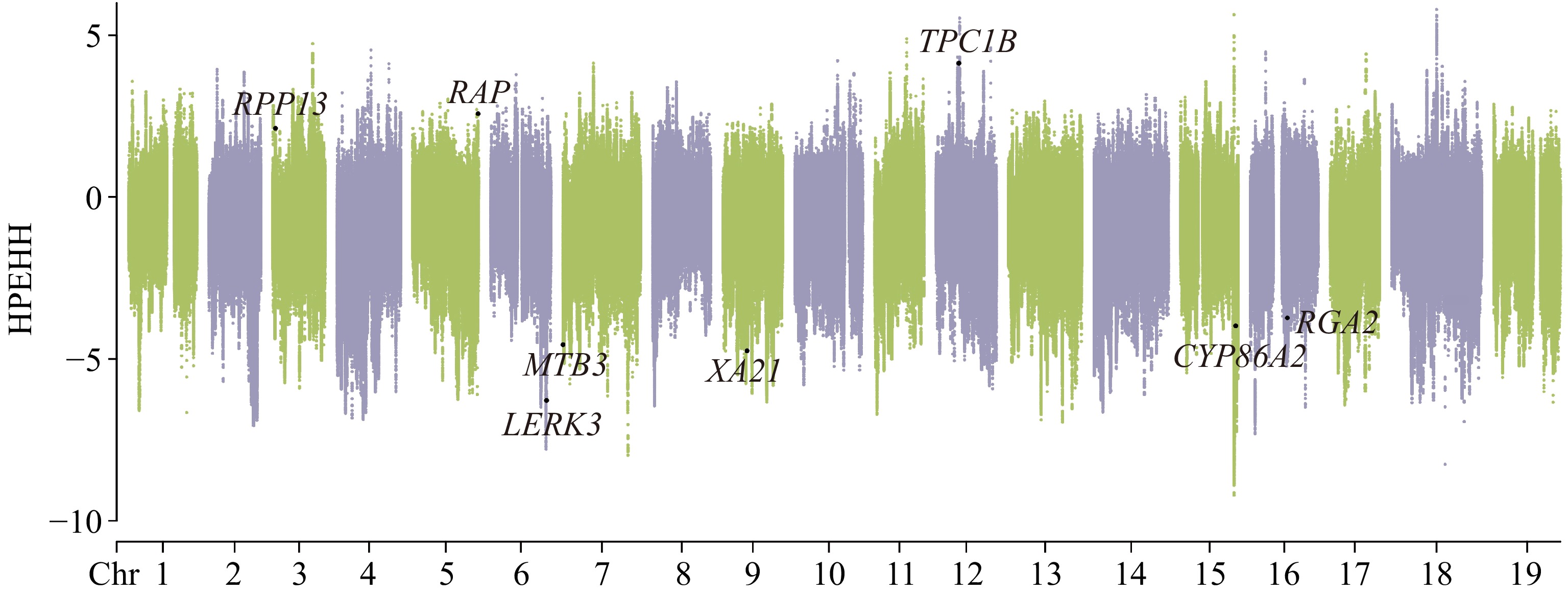
Figure 3.
Selective signal detected by XP-EHH analysis. V. adenoclada was taken as the candidate group and 'Shine Muscat' was regarded as the reference.
Positively selected genes in V. adenoclada and 'Shine Muscat' populations were then extracted respectively. There were 364 genes annotated to biological process (BP) terms for V. adenoclada, and 1,558 genes for 'Shine Muscat'. Five GO terms in V. adenoclada, including regulation of innate immune response (GO:0045088), jasmonic acid and ethylene-dependent systemic resistance (GO:0009861), DNA integration (GO:0015074), RNA-dependent DNA biosynthetic process (GO:0006278), DNA recombination (GO:0006310), were significantly enriched (Supplemental Fig. S1). By contrast, the GO terms enriched in 'Shine Muscat' included DNA integration (GO:0015074), RNA-dependent DNA biosynthetic process (GO:0006278), DNA recombination (GO:0006310), defense response (GO:0006952), and phytochelatin biosynthetic process (GO:0046938).
Disease resistance is one of the major agriculture indicators that are responsible for the steady crop yield[31]. More genes associated with disease resistance were under selection in 'Shine Muscat', and some distinct defense-response-related genes were found in both MUS and ADE groups. Several disease-resistant genes, like RPS4B, Xa21, MYC2, and MTB, were in the selected regions of 'Shine Muscat'. In plants like Arabidopsis thaliana, the RPS4B gene is a part of a paired resistance (R) gene system, such as RRS1B/RPS4B. This gene pair specifically recognizes the bacterial effector AvrRps4, highlighting its role in plant immunity[32]. Xa21 is a prominent disease-resistance gene in rice, encoding a receptor-like kinase critical for defense against bacterial blight[33]. In tomato, the transcription factor MYC2 not only activates jasmonate (JA)-responsive genes but also terminates JA signaling through the activation of MYC2-targeted BHLH1 (MTB1), MTB2, and MTB3 proteins. MTB proteins negatively regulate JA-mediated responses by disrupting the MYC2-MED25 complex, forming an autoregulatory negative feedback loop[34].
In V. adenoclada, some disease-resistance associated genes were also found, such as SUMM2, RPP13, RLP30, and EIX1, that were under positive selection. SUMM2 plays a role in counteracting the suppression of basal resistance by microbial effectors[35]. RPP13 safeguards the plant against pathogens carrying a specific avirulence protein through an indirect interaction with the alleged avirulence protein[36]. RLP30 is a receptor for microbe-associated molecular patterns (MAMPs) that triggers a BAK1-dependent basal immune response to necrotrophic fungi, such as Sclerotinia sclerotiorum[37]. And EIX1 was involved in plant defense, which confers resistance against the fungal pathogen T.viride through recognition of the EIX elicitor protein[38].
Additionally, the selected genes from V. adenoclada were specifically enriched in two KEGG pathways, biosynthesis of secondary metabolites and sesquiterpenoid and triterpenoid biosynthesis. Various secondary metabolites, such as flavonoids, and terpenoids, were reported to be associated with the abiotic stress response in plants (Supplemental Fig. S2)[39,40]. Given the wild environment that the V. adenoclada habitats, it is reasonable that these genes and pathways were positively selected by the harsh environment with multiple abiotic stress.
Characterization of genetic load
-
The genetic burden of 28 individuals was investigated using SIFT4G software. The number of heterozygous alleles, homozygous alleles that carry deleterious SNPs, and the overall number of deleterious SNPs of each group were recorded respectively (Fig. 4). We found that wild individuals V. adenoclada showed a significantly higher number of deleterious mutations than cultivated individuals 'Shine Muscat' (Fig. 4c). The total number of deleterious mutations in the hybrid individual HYB1 was higher than the 'Shine Muscat' individuals but lower than the V. adenoclada individuals. Specifically, compared with those parental species, the deleterious recessive alleles in the hybrid offspring HYB1 decreased substantially (Fig. 4b), while deleterious heterozygous alleles detected were higher than any of their parental accessions (Fig. 4a).
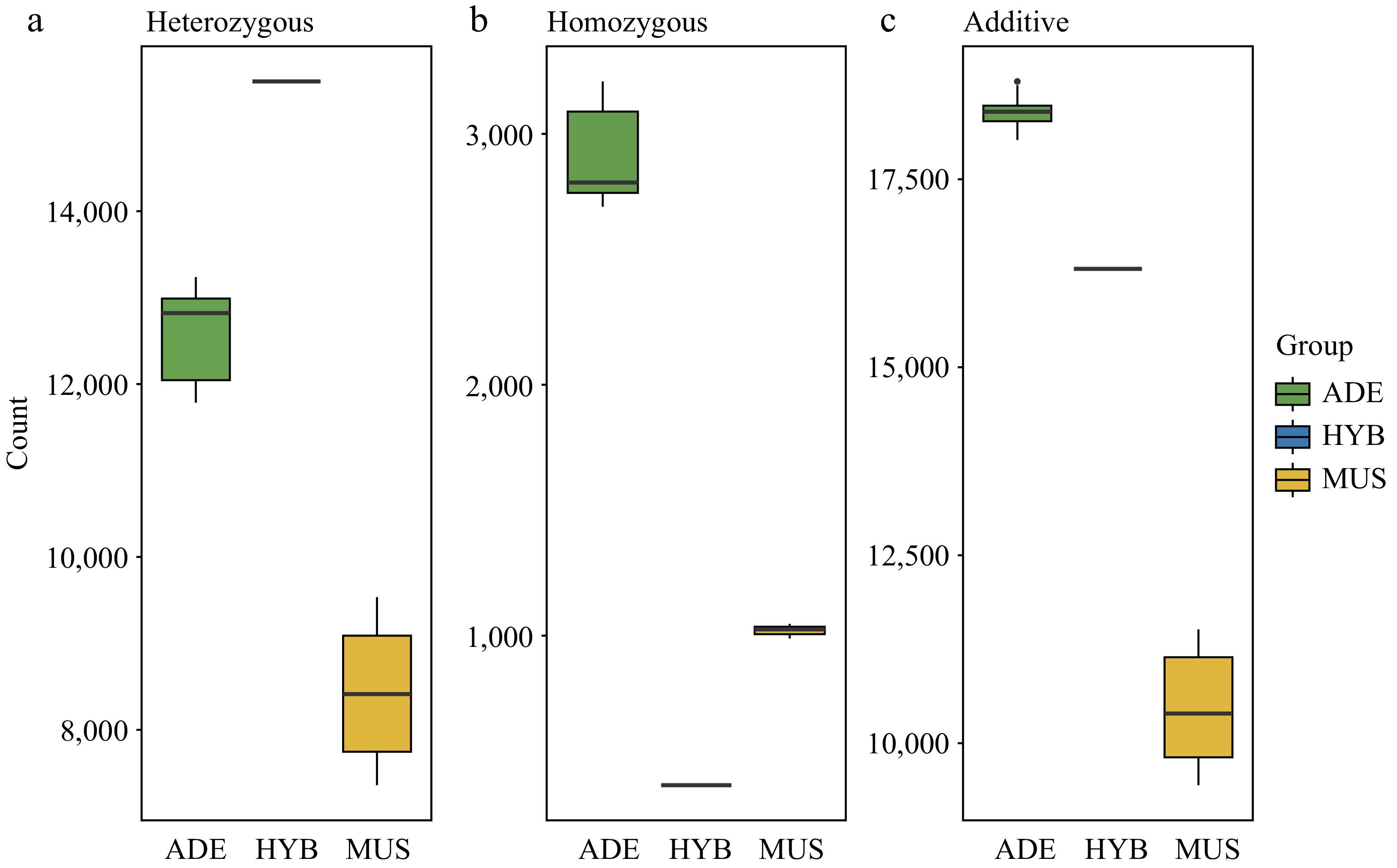
Figure 4.
Deleterious mutations analysis among V. adenoclada, 'Shine Muscat', and their one hybrid offspring. (a) The number of heterozygous deleterious alleles. (b) The number of homozygous deleterious alleles. (c) The total number of deleterious SNPs.
Then, corresponding genes with the sites that were considered homozygously deleterious within individuals of the ADE and MUS groups were utilized for enrichment analyses in each group, respectively. These two groups were both enriched for some of the identical GO terms, such as defense response (GO:0006952), protein phosphorylation (GO:0006468), DNA integration (GO:0015074), protein serine/threonine kinase activity (GO:0004674), ATP binding (GO:0005524), calmodulin binding (GO:0005516), and plasma membrane (GO:0005886) (Supplemental Fig. S3). The biological process, recognition of pollen (GO:0048544), was specifically enriched in the MUS group.
-
In this study, V. adenoclada and 'Shine Muscat' were clearly differentiated through phylogeny, admixture, and PCA analysis. Seven 'Muscat' individuals exhibited a single ancestral component in admixture analysis, while V. adenoclada manifested two main ancestries, which suggested the different sources of the V. adenoclada distributed in different regions. Three ADE individuals collected from Guangdong Province showed a mix of two ancestries, indicating that there might be gene flow of V. adenoclada in Guangxi and Guangdong Province, China. Besides, relative to the wild ADE_B population in Guangxi Province, the higher IBD score within the ADE_A group reflected the lower genetic diversity and closer kinship of these individuals. Combining genetic structure and kinship analysis, the HYB1 individual can be confirmed to be a progeny from the hybridization of V. adenoclada and 'Shine Muscat'.
Divergence of selection signal within V. adenoclada and Shine Muscat
-
The present analysis showed that there were many more loci under selection in 'Shine Muscat' compared with V. adenoclada individuals. At the same time, a lower number of SNPs was detected in 'Shine Muscat' (Table 1), which echoed such a result and suggested that many SNPs within the selected regions might be swept. This substantial difference in the number of selected loci between the two groups suggested the artificial selection and the breeding efforts applied to the cultivated 'Shine Muscat' and its parents[41].
Table 1. Quality and SNPs statistics of all 28 resequenced accessions.
Sample Total base (bp) Depth Mapping rate SNP number ADE1 17,520,817,445 35.40 97.32% 5,626,462 ADE2 24,257,947,082 49.02 88.66% 5,679,994 ADE3 18,328,442,529 37.04 80.59% 5,676,095 ADE4 21,011,413,063 42.46 94.82% 5,689,913 ADE5 22,031,567,287 44.52 97.23% 5,691,839 ADE6 24,068,277,235 48.64 96.30% 5,695,943 ADE7 23,436,537,148 47.36 96.31% 5,696,589 ADE8 21,950,203,360 44.36 96.11% 5,669,703 ADE9 22,002,259,768 44.46 96.97% 5,703,161 ADE10 22,309,202,148 45.08 96.37% 5,700,374 ADE11 22,049,567,098 44.56 96.66% 5,680,656 ADE12 27,580,752,978 55.73 97.70% 5,651,025 ADE13 22,064,551,844 44.59 97.65% 5,643,197 ADE14 24,850,211,192 50.22 97.83% 5,581,868 ADE15 22,814,344,130 46.10 97.74% 5,451,046 ADE16 23,240,185,342 46.96 97.54% 5,448,929 ADE17 19,291,800,581 38.98 98.07% 5,479,083 ADE18 19,298,624,750 39.00 97.67% 5,463,166 ADE19 20,634,991,493 41.70 97.95% 5,451,605 ADE20 20,619,496,304 41.67 98.06% 5,476,361 HYB1 19,995,439,567 40.41 94.71% 6,140,779 MUS1 20,104,866,097 40.63 97.08% 4,059,284 MUS2 31,473,565,455 63.60 98.29% 3,919,498 MUS3 32,273,677,414 65.22 98.88% 3,832,869 MUS4 35,479,482,412 71.69 98.91% 3,771,105 MUS5 35,076,902,316 70.88 97.71% 3,973,151 MUS6 36,508,316,389 73.77 98.92% 3,749,505 MUS7 7,963,746,000 16.09 98.68% 3,956,697 Corresponding to the selected situation around the whole genome, more selected genes related to disease resistance in 'Shine Muscat' were detected than in V. adenoclada. There are several specific resistance genes with distinct functions respectively in these two groups, which suggests their disparity with regard to the defense response. Besides, several genes associated with the pathway, biosynthesis of secondary metabolites, were specifically enriched in V. adenoclada. In light of the divergence exhibited in V. adenoclada and Shine Muscat, it is possible for the hybrid offspring to inherit the premium elements of each parent and become a new cultivar integrating the high resistance and good quality.
The accumulation of the deleterious mutation
-
Deleterious mutations could cause changes or loss of genetic functions, thereby potentially affecting the adaptation of the organism[19]. Genes have been detected with potentially deleterious mutations in both the ADE and the MUS groups, many of which were enriched in the same biological processes or molecular functions. This suggests a common impact of these mutations on the fitness of all grapes. The wild species V. adenoclada was found to consist of more deleterious mutations, both in the form of heterozygous alleles and homozygous alleles, than the 'Shine Muscat'. Some studies indicated that during the constant breeding period, many deleterious mutations with large effects would be purged by negative selection due to the exposure of recessive deleterious mutations when inbreeding[42]. In this case, cultivated 'Shine Muscat' individuals might have undergone the same process, though the wild ancestors of this cultivar need further consideration in future studies. The present study also investigated the accumulation of deleterious mutations in the hybrid offspring HYB1 and discovered that most of the deleterious variations were embedded in the heterozygous alleles. This phenomenon was consistent with a study about walnut cultivar improvement, which pointed out that during breeding, the deleterious mutations were converted from homozygous format to heterozygous format[43]. Some studies revealed an accumulation of moderately deleterious burden in the context of modern crop breeding, and showed that slightly deleterious mutations were more likely to continue to exist in the form of heterozygosity[18,42]. Combined with the current context of high heterozygosity in the hybrid offspring, it suggests that in the future, it will be necessary to figure out a way to target such deleterious mutations that are hidden within the heterozygous alleles and are detrimental to fitness, and eliminate them using modern molecular methods.
-
In this study, high-quality resequencing data was utilized from V. adenoclada, 'Shine Muscat', and their one hybrid offspring to investigate the genetic divergence between them. The present findings revealed distinct genetic structures between V. adenoclada and 'Shine Muscat' accessions and confirmed the hybrid origin of HYB1. A marked genetic divergence was observed, with the cultivated variety 'Shine Muscat' showing stronger selection signals across the genome. In terms of disease resistance, different biological processes, and genes under selection were enriched in wild and cultivated accessions. V. adenoclada exhibited selection signals in specific pathways, like the biosynthesis of secondary metabolites and sesquiterpenoid and triterpenoid biosynthesis, suggesting their potential adaptability to abiotic stress in the wild. Genetic load analysis highlighted that deleterious mutations in 'Shine Muscat' might have been mitigated through breeding, and it also indicated the importance of monitoring heterozygous deleterious mutations during the crossing experiment. However, further field experiments are required to investigate the potential influence of genetic load in hybrid offspring. This study delineates clear genetic differentiation between V. adenoclada and 'Shine Muscat', suggesting the potential to develop superior hybrids from grapevines with various premium traits.
-
The authors confirm contribution to the paper as follows: study conception and design: Peng W, Liang F, Chen Z; data collection: Wu D; analysis and interpretation of results: Peng W, Liang F, Zhang T; draft manuscript preparation: Peng W, Liang F, Chen Z; writing-review and editing: Wu D, Xu X, Ma Z, Pan F, Wei R, Li H, Gong Z, Yang X, Zhou Y; visualization: Peng W, Zhang M; supervision, Wu D, Xu X; project administration: Wu D; funding acquisition: Wu D. All authors reviewed the results and approved the final version of the manuscript.
-
The raw data that support the findings of this study are available in the NCBI repository. Raw data of accessions ADE1−ADE20, MUS1, and HYB1 that are newly sequenced in this study are stored under the BioProject of PRJNA1082482; Raw data of the accessions MUS2-MUS6 downloaded from the NCBI were in the number of DRR186531, DRR186532, DRR186533, DRR186535, DRR385775, respectively; and raw data of the MUS7 accession are not yet publicly available due to its source from the project under study. All necessary figures and tables generated or analyzed during this study are included in this published article and its supplementary information file.
This research was funded by Breeding and Application of Excellent and Characteristic Germplasm of Mao Grape, grant number 2018AB51020; Guangxi Luocheng Mao Grape Experimental Station Project (Grant No. Gui TS202118); Guangxi Bama County Science and Technology Talent Plan Project (Grant No. Barenke 20210039).
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Wenjing Peng, Feicui Liang, Zhuyifu Chen
- Supplemental Table S1 IBD scores of all 28 Resequenced accessions.
- Supplemental Fig. S1 GO enrichment plot of XP-EHH analysis for ADE and MUS groups. (A) GO enrichment with selected genes in ADE group. (B) GO enrichment with selected genes in MUS group.
- Supplemental Fig. S2 KEGG enrichment plot of XP-EHH analysis for ADE and MUS groups. (A) KEGG enrichment with selected genes in ADE group. (B) KEGG enrichment with selected genes in MUS group.
- Supplemental Fig. S3 GO enrichment plot of genetic load for ADE and MUS groups. (A) GO enrichment with potentially deleterious genes in ADE group. (B) GO enrichment with potentially deleterious genes in MUS group.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Peng W, Liang F, Chen Z, Gong Z, Zhang M, et al. 2024. Genomic signals of divergence and hybridization between a wild grape (Vitis adenoclada) and domesticated grape ('Shine Muscat'). Fruit Research 4: e028 doi: 10.48130/frures-0024-0022 

Genomic signals of divergence and hybridization between a wild grape (Vitis adenoclada) and domesticated grape ('Shine Muscat')
- Received: 04 March 2024
- Revised: 15 April 2024
- Accepted: 27 May 2024
- Published online: 05 August 2024
Abstract: Interspecific hybridization in Vitis holds immense potential for combining valuable adaptive traits and breeding superior cultivar development. To evaluate the feasibility of hybrid breeding using the resilient wild species Vitis adenoclada and the commercially successful cultivar V. vinifera 'Shine Muscat', their signatures of hybridization, genetic divergence, and divergent selection were investigated. Analyses of 28 resequencing genomes revealed pronounced genetic differentiation between these two lineages and corroborated the hybridization event within a derived progeny. Notably, 'Shine Muscat' exhibited stronger genome-wide selection signals, reflecting its intensive breeding history. While divergent selection signatures associated with disease resistance were evident in both species, V. adenoclada displayed enrichment in pathways linked to abiotic stress resistance. Furthermore, while 'Shine Muscat' displayed potentially mitigated deleterious mutations compared to V. adenoclada, their hybrid offspring exhibited an accumulation of heterozygous deleterious alleles, emphasizing the crucial need for monitoring such mutations in future breeding endeavors. Collectively, the findings unveil the significant genetic divergence and contrasting adaptations between V. adenoclada and 'Shine Muscat', highlighting their immense potential for breeding next-generation cultivars with enhanced resilience and superior quality.
-
Key words:
- Vitis adenoclada /
- 'Shine Muscat' /
- Hybridization /
- Genetic divergence /
- Genetic load