









-
The genus Artemisia, comprising small herbs and shrubs, contains over 500 species distributed worldwide[1]. As the largest genus in the family Asteraceae, many Artemisia species have been traditionally used as medicinal plants. They contain abundant secondary metabolites including volatiles, flavonoids, organic acids, and lactones, and have shown therapeutic potential for treating diseases such as malaria, hepatitis, cancer, and inflammation[2,3].
Most Artemisia species produce intense aromas due to their volatile organic compounds (VOCs) abundance. These VOCs can effectively deter herbivores and inhibit pathogenic microbes, conferring a selective advantage on plants[3]. Numerous studies have identified the insecticidal and antimicrobial potential of essential oils and extracts from the Artemisia species[4]. The essential oil of Artemisia lavandulaefolia exhibited contact and fumigant toxicity against Plutella xylostella adults[5]. The methanol leaf extracts and essential oil of Artemisia annua significantly reduced the body weight of cotton bollworm larvae at 2% concentration and strongly inhibited the growth of Fusarium oxysporum and F. solani[6,7]. However, the bioactivity of plant volatiles depends on genotype, tissue, environment, types of pests and diseases, etc. Therefore, more Artemisia species should be identified and evaluated for their insecticidal and antimicrobial potential assay. Exploring Artemisia germplasm diversity is critical for discovering novel bioactive compounds and developing biopesticides. Furthermore, Artemisia can provide potential parents for breeding resistant varieties of closely related species such as chrysanthemum.
Chrysanthemum (Chrysanthemum morifolium), one of the four traditionally famous flowers in China and the second most essential cut flowers globally, is of extremely high ornamental and economic value. Chrysanthemum is vulnerable to various pests and pathogens. The chrysanthemum aphid, Macrosiphoniella sanborni, is the most damaging pest, which not only hinders vegetative growth but also impacts the ornamental quality of flowers[8]. For pathogens, Alternaria alternata, Colletotrichum siamense, Phoma sp., and Fusarium solani are the primary pathogens of chrysanthemums that infect the aerial part or roots, causing chrysanthemums to wither, rot, and even die. At present, the control of these pests and diseases mainly relies on the use of chemical pesticides, which usually lead to environmental pollution, pesticide residue, and produce a negative impact on human health. As a related species of chrysanthemum, many Artemisia species are reportedly resistant to aphids and pathogens. For example, the essential oils from Artemisia monosperma strongly inhibited the mycelium growth of A. alternata and F. solani[9]. The present researchers previously found that Artemisia vulgaris, Artemisia japonica, Artemisia scoparia, and A. annua showed a strong repellent and antifeedant effect after inoculating aphids[10]. An intergeneric hybridization between aphid-susceptible Chrysanthemum 'Zhongshanjingui' (maternal parent) and A. vulgaris (paternal parent) showed much higher resistance to chrysanthemum aphid than maternal chrysanthemum[11]. Therefore, Artemisia plants have broad prospects in integrated pest and disease control and provide materials and new strategies for chrysanthemum green control and the breeding of pest or pathogen-resistant chrysanthemum cultivars.
The four Artemisia species, including A. keiskeana, A. viridisquama, A. maximowicziana, and A. sacrorum, distributed widely in China, are traditional Chinese medicinal plants used to treat many diseases (
www.iplant.cn ). Current research mainly focuses on their pharmacological activities, while their biological activities against pathogens and pests of plants are rarely reported[12−16]. This study aimed to evaluate the aphid resistance and antimicrobial properties of extracts from four Artemisia species. The repellent effects against aphids (M. sanborni) were assessed using choice and no-choice experiments, compared to C. morifolium 'Jinba'. The antimicrobial activity of ethyl acetate extracts of their different tissues was compared using the disc diffusion method against four selected pathogenic microorganisms. To identify potential active components, we analyzed the extracts by gas chromatography-mass spectroscopy (GC-MS) and then evaluated them using the OPLS-DA method. -
A. keiskeana, A. viridisquama, A. maximowicziana, A. sacrorum, and C. morifolium 'Jinba' were obtained from the Chrysanthemum Germplasm Resource Preserving Center, Nanjing Agricultural University, Nanjing, China. All plants were propagated from cuttings and grown in a greenhouse at 25 ± 2 °C, 70% relative humidity, and a 16 h/8 h light/dark photoperiod. The light intensity was 100 μmol·m−2·s−1. One-month-old plants were used for the following experiments.
Aphid performance assay on Artemisia species and C. morifolium 'Jinba'
-
Aphids reared on C. morifolium 'Jinba' at 25 ± 2 °C, 70% relative humidity, and a 16 h/8 h (light/dark) photoperiod was used. Aphid performance on Artemisia species and C. morifolium 'Jinba' were tested using a no-choice assay according to previous methods with slight modification[10]. Each assay contained 12 individual plants with three biological replications. Ten adult aphids were inoculated on the terminal bud of plants after 4 h starvation. Then, each plant was placed in a 25 cm × 12 cm polyester cylinder capped with gauze. Total aphid numbers were counted every other day.
Aphid olfactory bioassay
-
Aphid olfactory responses were measured using a Y-tube olfactometer (25 cm × 15 cm × 15 cm, 1 cm diam, 75° angle) based on previous methods with slight modification[17]. The arms were connected by Teflon tubes to chambers containing the odor sources. Charcoal-filtered air was pumped through each chamber at a velocity of 100 mL·min−1. Adult aphids starved for 4 h were placed at one end of the Y-tube. The aphids that moved into one of the arms up to 3 cm and stayed over the 30 s were recorded as making an effective choice, and those that did not choose within 5 min were deemed as 'no choice'. Each assay contains ten individual aphids with four biological replications. The Y-tube was rotated 180° to change the odor source every 5 min. The olfactometer tube was washed with 70% ethanol and dried at 100 °C for 5 min.
Volatiles collection and analysis
-
The second to fourth leaves from the plant apex, stems, and roots were harvested for volatile extraction. 200 mg of tissue was ground in liquid nitrogen and extracted with 1 mL ethyl acetate containing 0.002% (w/v) nonyl acetate as an internal standard. The extraction was performed under shaking at 200 rpm for 1 h at room temperature. After centrifugation at 12,000 rpm for 15 min, the supernatant of extracts was collected and stored at −80 °C for further analysis and the antimicrobial assay in vitro.
The extracts were analyzed by GC-MS (Agilent 9000 B-7000D) and equipped with an HP-5 MS capillary column (30 m × 0.25 mm × 0.25 μm film thickness). The GC procedure was set as follows: 1 μl of each sample was injected at 250 °C with 5:1 split mode. The oven temperature was increased at 5 °C·min−1 from 40 to 80 °C and held for 2 min, then increased to 160 °C at 5 °C·min−1 and held for 2 min, followed by increasing to 250 °C at 10 °C·min−1. The flow rate of helium carrier gas was set at 1 mL·min−1. All MS data was collected from 40 to 400 m/z.
The extracts' components were identified by matching retention time and mass spectra to libraries (National Institute of Standards and Technology, NIST-2008). The content of each component was calculated using the internal standard method. The calculation formula is as follows: the content of each component (μg·g−1 FW) = [(peak area of each constituent × content of internal standard)/peak area of internal standard]/sample fresh weight, where FW represents fresh weight.
Antimicrobial properties of the extracts from Artemisia plants
-
Four frequently occurring pathogenic microorganisms were employed, including A. alternata, C. siamense, Phoma sp., and F. solani, isolated from infected chrysanthemums. The in vitro antimicrobial activity of ethyl acetate extracts was evaluated by the agar diffusion method. Fungal plates cultured for five days were selected. Then, an agar mycelium block with a diameter of 0.6 cm was placed in the center of the potato agar medium with evenly spread 200 μL of extracts. 200 μL of ethyl acetate was used as the negative control. The plates were incubated at 28 °C for 4 d. The colony diameter of the fungi was measured with a caliper and photographed. Each treatment was repeated five times.
Antimicrobial activity was evaluated by calculating the inhibition percentage of colony diameter compared to the negative control as follows: Inhibition rate (IR) = (M − T)/M × 100, where M = diameter of negative control colonies and T = diameter of test colonies.
Statistical analysis
-
SPSS v 20.0 was used for all statistical analyses. Data were expressed as mean ± standard deviation (SD). Duncan's multiple range test (p < 0.05) and one-way analysis of variance were performed to determine the significance of the data. Orthogonal Partial Least Squares Discriminant Analysis (OPLS-DA) was conducted using SIMCA 14.1. GraphPad Prism 10 was used to cluster heat map analysis.
-
Aphid populations were monitored every other day for 7 d following the inoculation of ten adults onto each plant. Aphid populations on Artemisia spp. gradually decreased, reaching almost zero on the 7th day. However, the number of aphids on C. morifolium 'Jinba' gradually increased, reaching about 5.9 times on the 7th day (Table 1; Fig. 1). The results suggest that the tested Artemisia spp. exhibited apparent antifeedant effects on aphids compared to C. morifolium 'Jinba'.
Table 1. Identification of resistance of Artemisia species to aphids.
Plants No. of aphids at different days after inoculation Multiplication rate 1 d 3 d 5 d 7 d C. morifolium 'Jinba' 10.0 ± 0.00c 15.9 ± 2.15d 31.7 ± 5.43e 59.1 ± 7.29f 5.9 ± 0.73 A. keiskeana 10.0 ± 0.00c 2.6 ± 1.51b 0.6 ± 0.79a 0.0 ± 0.00a 0.00 ± 0.00 A. viridisquama 10.0 ± 0.00c 3.5 ± 1.73 b 0.4 ± 0.67a 0.0 ± 0.00a 0.00 ± 0.00 A. maximowicziana 10.0 ± 0.00c 3.3 ± 1.95 b 0.2 ± 0.63a 0.0 ± 0.00a 0.00 ± 0.00 A. sacrorum 10.0 ± 0.00c 2.7 ± 0.98b 0.1 ± 0.29a 0.0 ± 0.00a 0.00 ± 0.00 Values (given as mean ± SD) labelled with different letters represent significant differences (p < 0.01). 
Figure 1.
Aphid density on C. morifolium 'jinba' and four Artemisia species at 5 d after aphid inoculation.
Y-tube olfactometer bioassay
-
To determine the repellent effects of Artemisia spp. volatiles on aphids, dual-choice assays were performed using a Y-tube olfactometer. Each Artemisia species coupled with C. morifolium 'Jinba' as a pair of odor sources in the assay. Compared to C. morifolium 'Jinba', all tested Artemisia species elicited obvious avoidance responses to aphids (Fig. 2). In particular, A. sacrorum has the strongest repellent effect on aphids, followed by A. viridisquama.
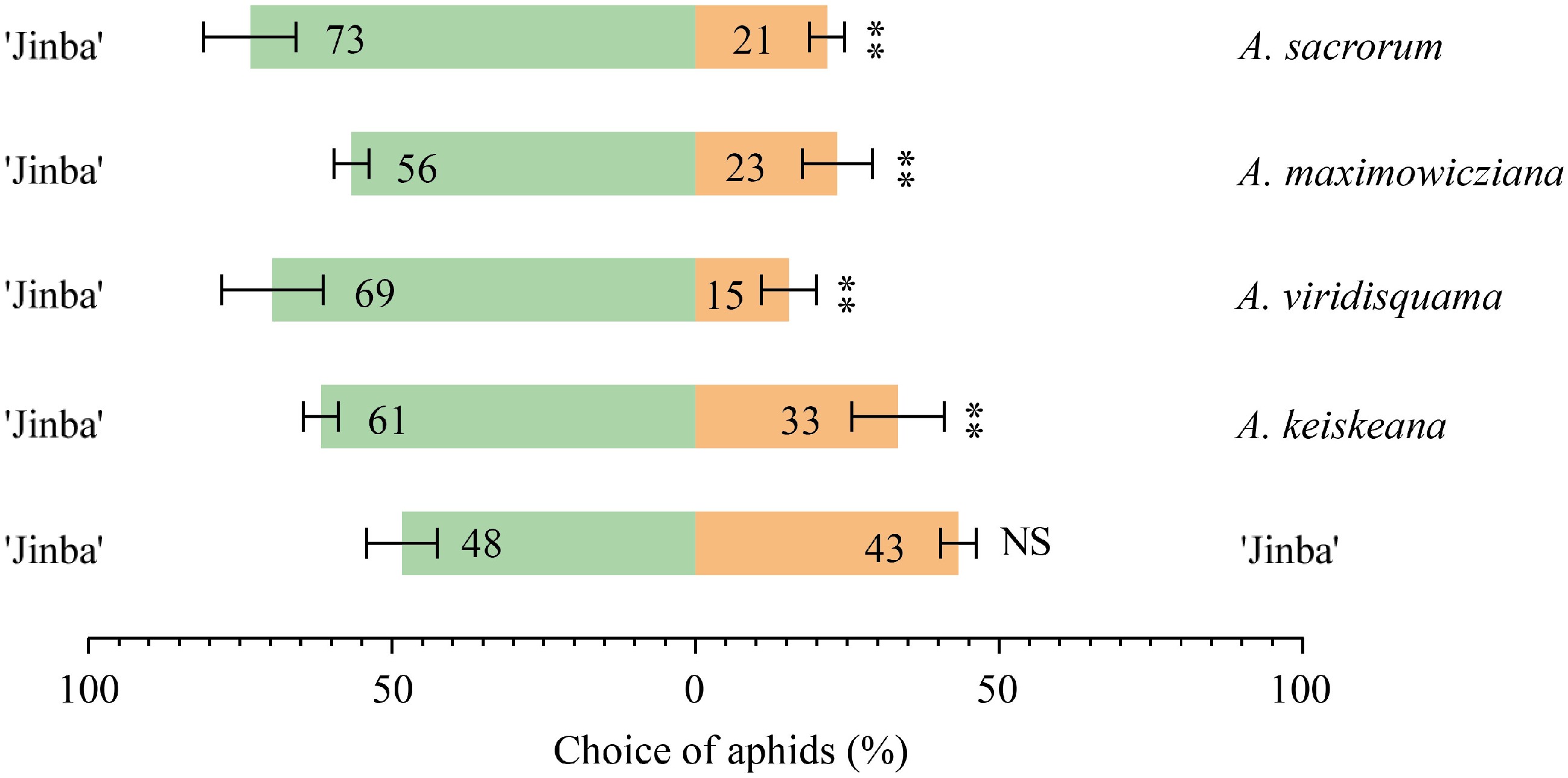
Figure 2.
Choices of aphid to Artemisia species and C. morifolium 'Jinba' in the Y-tube olfactometer. The asterisks with the choice bars indicate significant preference. **p < 0.01.
Antimicrobial activity
Antifungal effect of leaf and stem extracts on Alternaria alternata, Colletotrichum siamense and Phoma sp.
-
A. alternata, C. siamense, and Phoma sp. commonly infect aerial tissues including the leaf and stem of chrysanthemum. Therefore, to track antifungal agents of aerial tissues of Artemisia spp., leaf and stem extracts were prepared and tested for antifungal activity against the three fungi.
For inhibition activity assay of the extracts on A. alternata, leaf extracts of A. keiskeana and stem extracts of A. keiskeana, A. viridisquama, and A. maximowicziana strongly inhibited the growth of A. alternata with inhibition rates of 71.35%, 71.93%, 74.85%, and 75.44%, respectively. In comparison, A. sacrorum showed the weakest inhibition, with rates of 29.24% (Table 2; Fig. 3a).
Table 2. Inhibitory effect of volatiles from leaves and stems of four kinds of Artemisia on the growth of A. alternata, C. siamense, and Phoma sp.
Plants Tissue Inhibition rate (IR) A. alternata C. siamense Phoma sp. A. keiskeana L 71.35 ± 1.17 57.14 ± 6.08 40.00 ± 2.40 S 71.93 ± 1.50 35.71 ± 3.02 19.51 ± 3.72 A. viridisquama L 33.00 ± 6.43 25.82 ± 1.92 12.20 ± 2.18 S 74.85 ± 3.31 42.86 ± 6.87 20.00 ± 1.86 A. maximowicziana L 53.80 ± 3.80 54.95 ± 2.91 61.46 ± 2.84 S 75.44 ± 3.31 51.65 ± 7.14 79.51 ± 2.56 A. sacrorum L 29.24 ± 3.80 19.78 ± 7.93 13.66 ± 3.28 S 29.24 ± 2.34 38.00 ± 4.12 15.12 ± 1.86 Values were given as mean ± SD. L, leaf; S, stem. 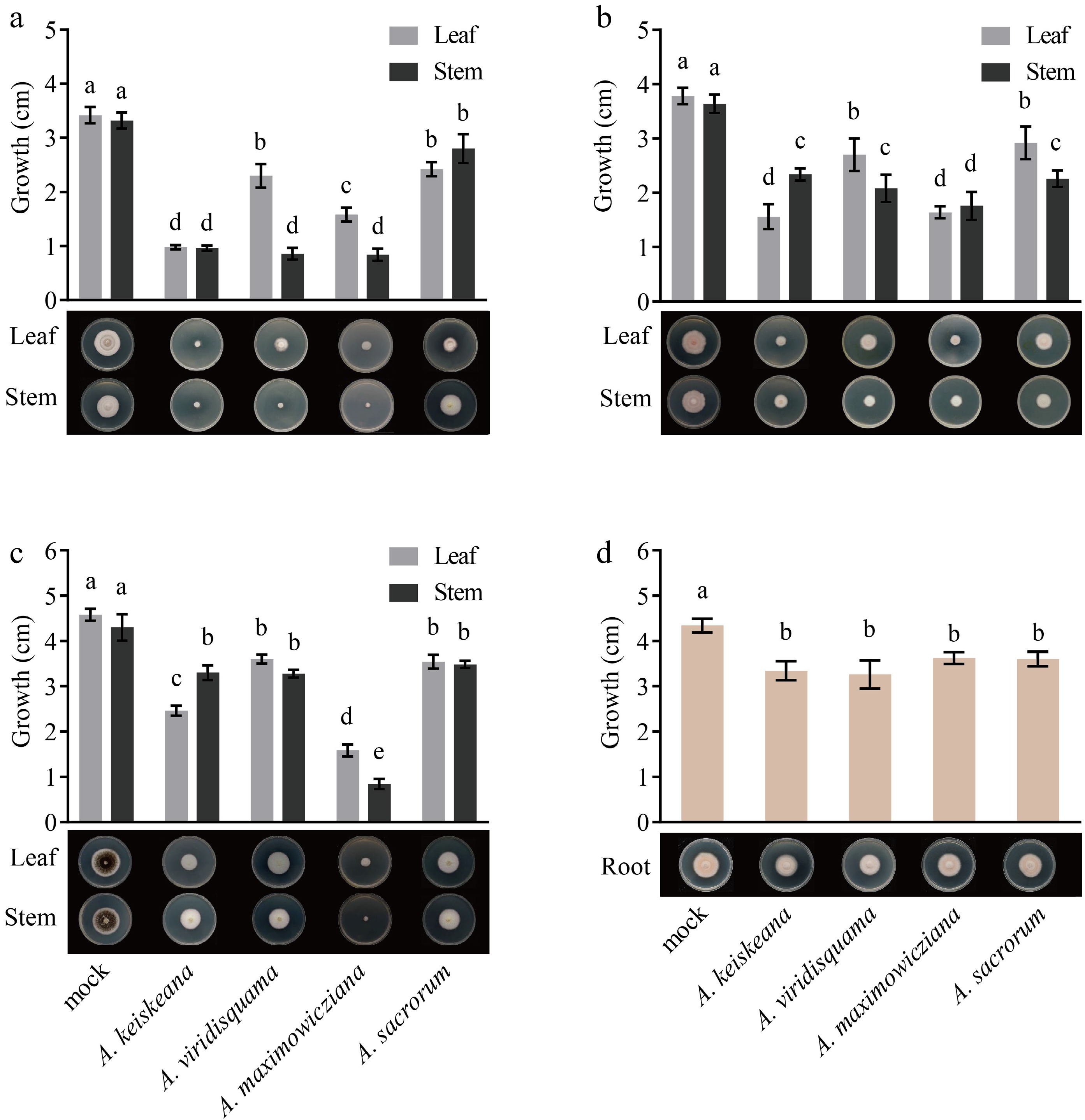
Figure 3.
Inhibitory effect of different tissue extracts from four Artemisia plants on mycelia growth. (a), (b) and (c) colony diameter (cm) of A. alternata, C. siamense, and Phoma sp. after treatment with leaf and stem extracts for 4 d, respectively; (d) growth (cm) of F. solani after treatment with root extracts for 4 d. Different letters represent significant differences (p < 0.05).
For inhibition activity assay of the extracts on C. siamense, the best inhibitory activity against C. siamense was found in leaf extracts of A. keiskeana and leaf and stem extracts of A. maximowicziana (57.14%, 54.95%, and 51.65%, respectively), followed by stem extracts of A. viridisquama, A. sacrorum, and A. keiskeana, with inhibitory rates of 42.86%, 38%, and 35.71%, respectively (Table 2; Fig. 3b).
For inhibition activity assay of the extracts on Phoma sp., leaf and stem extracts of A. maximowicziana showed superior inhibition, and the inhibitory rate was 61.46% and 79.51%, respectively. Stem extracts of A. keiskeana showed moderate inhibition with an inhibition rate of 40%. The rest had slight inhibition on Phoma sp. with rates below 20% (Table 2; Fig. 3c).
Antifungal effect of root extracts from Artemisia species on Fusarium solani
-
F. solani is the primary pathogen causing chrysanthemum root rot[18,19]. The antimicrobial activity of root volatiles of Artemisia spp. was tested against the mycelial growth of F. solani. The result indicated that these root extracts showed an inhibitory effect. Among them, the inhibitory activity of root extracts from A. keiskeana, A. viridisquama, A. maximowicziana, and A. sacrorum was 23.04%, 24.88%, 16.59% and 17.05%, respectively (Table 3; Fig. 3d).
Table 3. Inhibitory effect of volatiles from roots of four kinds of Artemisia on the growth of F. solani.
Plants Tissue Inhibition rate (IR) A. keiskeana R 23.04 ± 4.84 A. viridisquama R 24.88 ± 7.14 A. maximowicziana R 16.59 ± 2.99 A. sacrorum R 17.05 ± 3.68 Values were given as mean ± SD. R, root. Analysis of volatile compounds
-
To further investigate the potential bioactive components that confer resistance against aphids and pathogens, root, leaf, and stem extracts from Artemisia spp. were analyzed by GC-MS. Constituents were identified by matching mass spectra to the NIST library. Volatile content was quantified relative to an internal standard (Supplemental Tables S1−S4).
Nineteen compounds were identified in A. keiskeana (Supplemental Table S1). (−)-Alpha-pinene and camphene occurred across all tissues, with the highest levels in leaves followed by that in stems. The major leaf volatiles were (+)-2-bornanone (126.3 μg·g−1 FW), caryophyllene (135.6 μg·g−1 FW), and phytyl acetate (205.6 μg·g−1 FW). (−)-Beta-elemene (109.2 μg·g−1 FW) and aromadendrene (101.4 μg·g−1 FW) predominated in the roots (Supplemental Table S1).
Fifteen volatiles were found in A. viridisquama (Supplemental Table S2). Phytyl acetate (221.0 μg·g−1 FW), eucalyptol (47.1 μg·g−1 FW) and neophytadiene (67.1 μg·g−1 FW) were most abundant.
Twenty-five compounds were detected in A. maximowicziana, including six bioactive oxidized monoterpenes like (E)-thujone and (−)-thujol (Supplemental Table S3). (E)-thujone and (−)-thujol were the main components in leaf and stem extracts, the content of which in leaves was up to 174.4 μg·g−1 FW and 498.7 μg·g−1 FW, respectively.
A. sacrorum contained 26 volatile compounds (Supplemental Table S4), dominated by monoterpenes, sesquiterpenes, and diterpenes. (+)-2-Bornanone (193.3 μg·g−1 FW) and (−)-zingiberene (131.2 μg·g−1 FW) predominated in leaves.
Overall, 43 volatiles were detected across all Artemisia species (Fig. 4), and terpenoids were the predominant volatiles across all species and tissues. Camphene, gamma-elemene, phytyl acetate, and caryophyllene occurred in all species. Especially certain oxygenated monoterpenes (cis-4-thujanol, (E)-thujone, (−)-thujol, etc.) were unique to A. maximowicziana. Some sesquiterpenes, like (−)-beta-elemene, are only found in the root extracts of A. keiskeana. However, which components of these extracts played a vital role in the antifungal activity remains to be further analyzed.
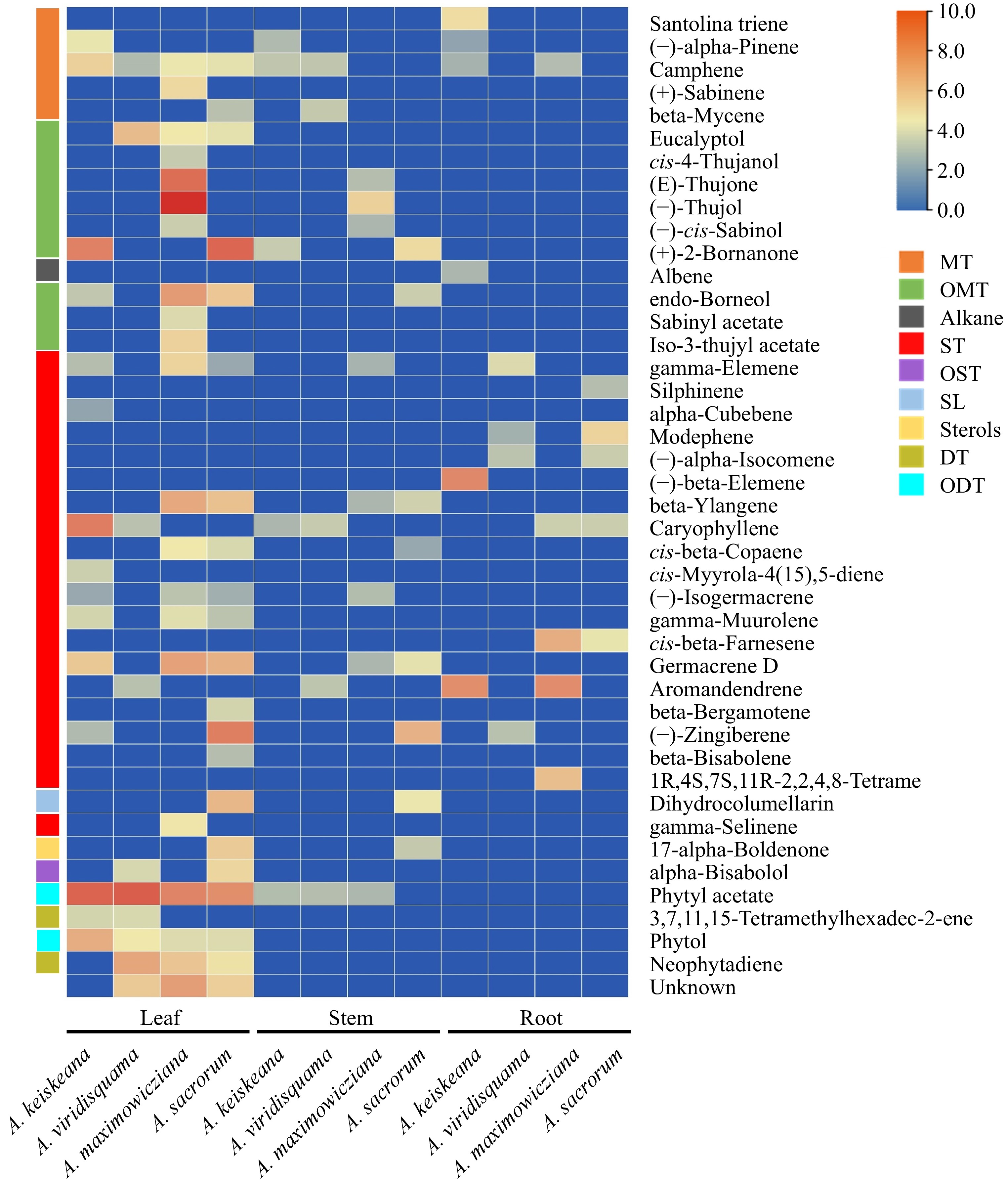
Figure 4.
Main components (above 1% of total volatiles present in chromatograms) of ethyl acetate extracts from the leaf, stem and root of four Artemisia species. Colors reflect the VOC's average relative content, n = 3. MT: monoterpenes; OMT: oxygenated monoterpenes; ST: sesquiterpenes; OST: oxygenated sesquiterpenes; SL: sesquiterpene lactones; DT: diterpenes; ODT: oxygenated diterpenes.
Screening of key bioactive components of extracts with antifungal effect
-
The contribution of each volatile compound of extracts to antimicrobial activity was assessed by Orthogonal Partial Least Squares Discriminant Analysis (OPLS-DA) based on the variable's importance in the project (VIP). VIP values were used to describe the overall contribution of each variable to the model. VIP ≥ 1 commonly used as the screening criterion showed that the variable has a significant influence on the model. Among the 35 components in stem and leaf extracts from four Artemisia species, 12 compounds including (−)-thujol, (+)-2-bornanone, phytyl acetate, (−)-zingiberene, (E)-thujone, unknown (unidentified compound), germacrene D, caryophyllene, endo-borneol, beta-ylangene, dihydrocolumellarin and neophytadiene, the VIP values of which were greater than 1, inhibited the growth of A. alternata hypha (Fig. 5a). Ten components-(−)-thujol, (E)-thujone, phytyl acetate, (+)-2-bornanone, endo-borneol, unknow (unidentified compound), germacrene D, caryophyllene, (−)-zingiberene and beta-ylangene contributed to the inhibition of C. siamense mycelial growth (Fig. 5b). For Phoma sp., the mycelial growth was inhibited by 12 key active compounds including (−)-thujol, phytyl acetate, (+)-2-bornanone, caryophyllene, (E)-thujone, (−)-zingiberene, germacrene D, neophytadiene, unknown (unidentified compound), endo-borneol, phytol, and beta-ylangene (Fig. 5c). The primary antifungal agents in the extracts varied among different pathogens, with (−)-thujol exhibiting the most potent influence. Regarding soil-borne pathogenic fungi F. solani, (−)-beta-elemene, aromandendrene, cis-beta-farnesene, 1R,4S,7S,11R-2,2,4,8-tetrame, santolina triene, modephene and caryophyllene from root extracts were effective constituents that could inhibit the growth of the hypha of F. solani, of which (−)-beta-elemene showed the greatest inhibitory effect on F. solani (Fig. 5d).
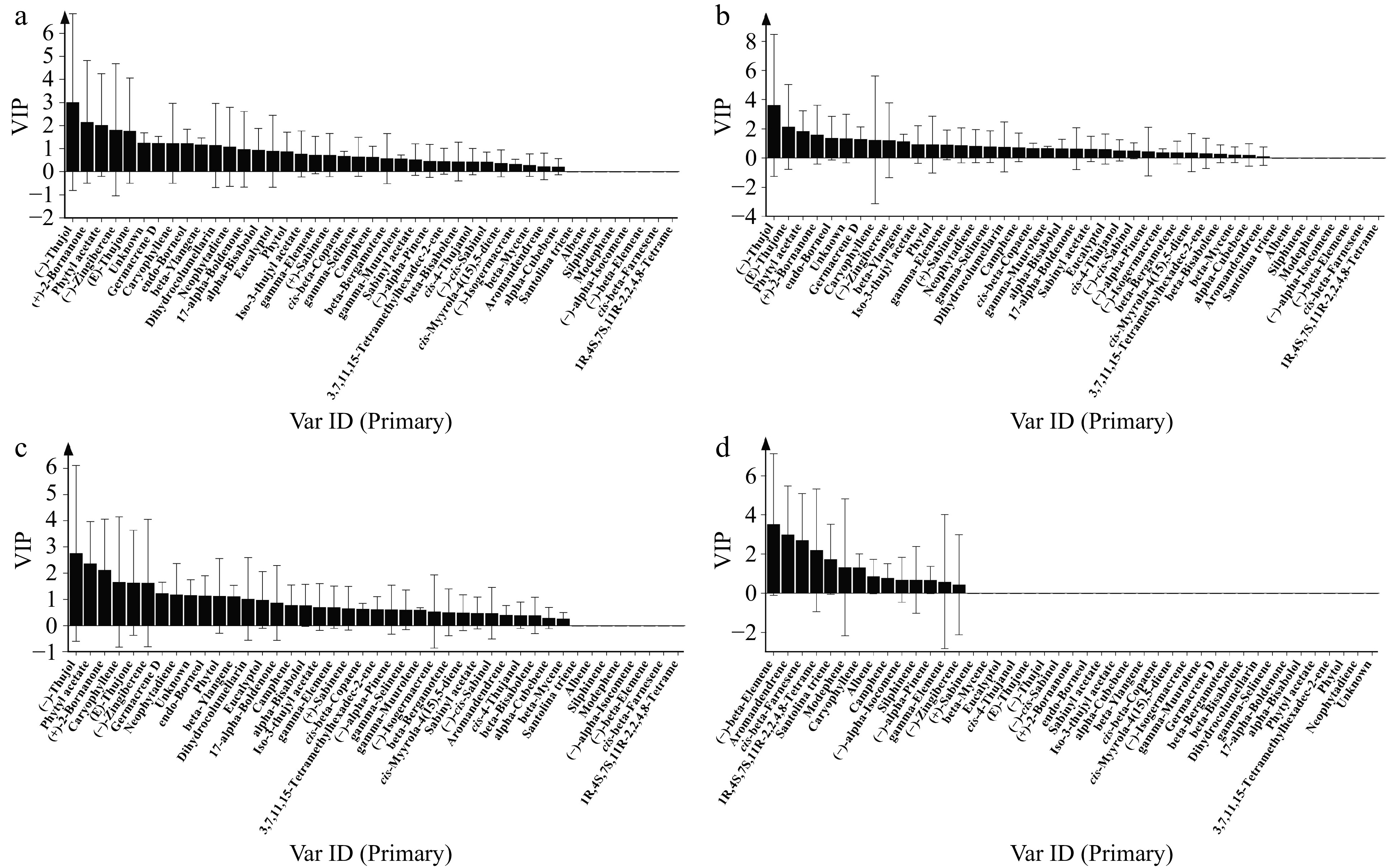
Figure 5.
Screening of key bioactive components of extracts with antifungal effect against (a) A. alternata, (b) C. siamense, (c) Phoma sp. and (d) F. solani by OPLS-DA. VIP ≥ 1 was used as the screening criterion.
-
Artemisia species are of great value due to their abundant bioactive compounds with pharmaceutical and pesticide applications. They also serve as parent materials for intergeneric hybrids with crops like chrysanthemum. Here, the aphid (M. sanborni) resistance of four Artemisia species and the antimicrobial properties of their extracts were investigated, to screen out excellent germplasm materials and active compounds.
Plant volatiles defend against aphids in direct and indirect ways, the former usually by direct contact or fumigation to repel and keep them from feeding, and even be toxic[20,21]. The essential oils of Artemisia monosperma were the most potent toxicants to Aphis nerii, displaying an LC50 value of 0.06 mg·L−1[22]. Sesquiterpenes from Solanum habrochaites have been verified to not only have a direct avoidance or anti-feeding effect on potato aphids but also affect their longevity and fecundity[23,24]. In this study, the four Artemisia species tested contained abundant bioactive terpenoids like caryophyllene, beta-farnesene, beta-bergamotene, beta-bisabolene, cis-4-thujanol, etc., known to deter aphids[23,25,26], likely contributing to the observed antifeeding and repellent effects. It suggests that all four Artemisia species are promising aphid-resistant germplasm with prospective bioactive volatiles.
The antifungal activity of Artemisia extracts depends on the extraction solvent, plant part, and pathogen species. Antifungal effects of A. annua leaf methanol extracts on F. oxysporum and F. solani increased with concentration, whereas root and stem extracts showed no inhibition[7]. Artemisia proceriformis oil more strongly inhibited Septoria glycines and Septoria tritici (MIC100 = 2.7 mg·mL−1) versus Fusarium graminearum and Fusarium verticillioides (MIC100 = 10.6 mg·mL−1)[27]. Here, differences in the antifungal activity of Artemisia extracts were also found. A. keiskeana leaf and stem extracts inhibited A. alternata over 70% but had minimal effects on Phoma sp. (Table 2). The inhibitory effect of stem extract from A. viridisquama on A. alternata was significantly higher than that of leaf extract. Moreover, we also noticed that A. maximowicziana extracts strongly inhibited all three aerial pathogens by over 50%. Thus, it represents a promising broad-spectrum botanical fungicide candidate.
GC-MS profiling of volatiles revealed that, as expected, terpenoids dominated the volatile profile, aligning with findings in other Artemisia species[1]. It mainly consisted of 1, 8-cineole, beta-pinene, thujone, artemisia ketone, camphor, caryophyllene, camphene, and germacrene D[4]. However, there were great differences in main compounds with some special species. For example, A. maximowicziana studied in this paper, whose major component was (−)-thujol, contained about three times as much as (E)-thujone. In addition, phytyl acetate, which was not mentioned in the previously reported Artemisia, was found to have a high content in all four tested Artemisia species.
Different pests and pathogens have different susceptibility to varied active compounds. The essential oil and extracts of Artemisia absinthium have demonstrated anti-settling, antifeedant and antimicrobial activity against various pests and pathogens such as Myzus persicae, Spodoptera littoralis, Tetranychus cinnabarinus, Diaphorina citri, Fusarium spp. and Botrytis cinerea[28−31]. Specifically, (−)-cis-chrysanthenol and linalool have been identified as the primary antifungal agents of extracts against Fusarium spp. and B. cinerea[29], while carvacrol, (−)-α-bisabolol and chamazulene were toxic constituents of volatile oils against D. citri[29,31]. In this study, the antifungal effects of (−)-thujol were potentially broad spectrum. For root pathogens, L-camphor, DL-camphor, β-caryophyllene, and camphene were reported to have antifungal efficacy against F. oxysporum and F. solani comparable to the synthetic fungicides flutriafol, and hymexazol[7]. Here, however, the VIP value of camphene was less than 1; probably, its content in the root was too low to be effective for the resistance. Based on previous reports and our findings, the four Artemisia plants contained 15 bioactive constituents-(−)-thujol, (+)-2-bornanone, phytyl acetate, (−)-zingiberene, (E)-thujone, unknown (unidentified compound), germacrene D, caryophyllene, endo-borneol and beta-ylangene, camphene, cis-beta-farnesene, beta-bergamotene, beta-bisabolene, cis-4-thujanol—may mediate the antifungal and anti-insect effects. However, the active ingredients individually and in combinations against aphids and the tested pathogens should be further studied.
-
In conclusion, the four Artemisia species tested showed certain repellent and antifeedant effects on aphids, and antifungal effects on A. alternata, C. siamense, Phoma sp., and F. solani. Their volatile extracts contain multiple bioactive compounds with the potential to be biofungicides. These findings provide a material and theoretical basis for developing new biopesticides and breeding of pest- and disease-resistant chrysanthemum cultivars.
-
The authors confirm contribution to the paper as follows: study conception and design: Chen S, Chen F; data collection: Yang M, Li M; analysis and interpretation of results: Yang M, Li M; draft manuscript preparation: Yang M. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
This work was supported by the National Natural Science Foundation of China (32372749), Hainan Provincial Natural Science Foundation of China (323CXTD386), the Program for Key Research and Development, Jiangsu, China (BE2023367, BE2022417), A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, PAPD. We thank Dr. Yuehua Ma (Central Laboratory of College of Horticulture, Nanjing Agricultural University) for assistance in using GC-MS (Agilent 9000 B-7000D).
-
The authors declare that they have no conflict of interest.
- Supplemental Table S1 Components in leaf, stem and root of A. keiskeana.
- Supplemental Table S2 Components in leaf, stem and root of A. viridisquama.
- Supplemental Table S3 Components in leaf, stem and root of A. maximowicziana.
- Supplemental Table S4 Components in leaf, stem and root of A. sacrorum.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yang M, Li M, Chen F, Chen S. 2024. Bioactive components and antimicrobial potential of extracts from Artemisia species and their repellent activities against Aphid (Macrosiphoniella sanborni). Ornamental Plant Research 4: e025 doi: 10.48130/opr-0024-0021 

Bioactive components and antimicrobial potential of extracts from Artemisia species and their repellent activities against Aphid (Macrosiphoniella sanborni)
- Received: 18 March 2024
- Revised: 11 June 2024
- Accepted: 18 June 2024
- Published online: 02 September 2024
Abstract: Species from the Artemisia genus frequently have high resistance to pests and pathogens due to their being rich in secondary metabolites. Therefore, identifying bioactive components from Artemisia plants is essential for developing botanical pesticides and selecting parents for breeding resistant varieties of cultivated Chrysanthemum morifolium. This study investigated the resistance of four Artemisia species to aphids (Macrosiphoniella sanborni) and the antimicrobial properties of their extracts. Choice and no-choice assays showed that the tested four species had strong repellent and antifeedant effects on aphids compared with chrysanthemum. The antimicrobial activity of ethyl acetate extracts from different tissues against four pathogenic fungi was tested by disc diffusion assay. Among them, the extracts from Artemisia maximowicziana showed the strongest antimicrobial effect. The inhibition rates of Alternaria alternata, Colletotrichum siamense, and Phoma sp. caused by leaf extracts from A. maximowicziana were 53.8%, 54.95%, and 61.46%, respectively. And the inhibition increased to 75.44%, 51.65%, and 79.51%, respectively, using the stem extracts. However, the root extracts of Artemisia spp. showed only up to 25% to Fusarium solani. GC-MS analysis showed that the volatiles of Artemisia spp. were mostly abundant in terpenoids, but the components and contents were remarkably different among species. Further analysis of Orthogonal Partial Least Squares Discriminant Analysis (OPLS-DA) showed the most contributed component among all potentially antimicrobial bioactive components was (−)-thujol. In this study, A. maximowicziana was identified as the material with potential value as a parent for crossbreeding, and its primary volatile compound (−)-thujol with potential resistant active is worth further investigation.
-
Key words:
- Aphid /
- Pathogen /
- Chrysanthemum /
- Secondary metabolites /
- Artemisia