








-
Both larvae and queens are fed royal jelly, which is produced by the hypopharyngeal and mandibular glands of young honeybees (Apis mellifera) in the colony[1,2]. Royal jelly is fed to all larvae during the first three days of their development. After this short period, worker bees switch to their special diet of pollen, honey, and nectar, while queen larvae continue to consume large amounts of royal jelly throughout their adult lives[3,4]. This differential feeding produces two different female castes, a long-lived queen and a short-lived worker. Interestingly, the worker bees are smaller and functionally sterile, whereas the queen is the largest member of the colony and has fully developed ovaries[5−7]. This phenotypic polymorphism in female honeybees is generated from two identical genomes by diet-controlled epigenetic mechanisms, mainly DNMT3-mediated DNA methylation[3,8,9].
Inhibition of DNMT3 in larvae resulted in 72% of adult bees becoming queens with fully developed ovaries, similar to those of queens reared on pure royal jelly in the hive, suggesting that DNMT3 inhibition induces royal jelly-like effects on the caste phenotype of honeybees[8]. This suggests that one or more of the biologically active components of royal jelly may specifically inhibit DNMT3. Thus, one of the key questions to be addressed in the honeybee genome is to identify the epigenetically active compounds in royal jelly that inhibit DNMT3 and thereby determine developmental fate.
In addition to proteins, vitamins, mineral salts, lipids, enzymes, and carbohydrates, royal jelly contains small amounts of polyphenols, including derivatives of luteolin and kaempferol (Table 1), ranging from 14 to 18,936 μg/kg[1,10]. DNMT3 is a target of luteolin in its mechanistic action against human cancer cells[11,12] and of kaempferol in a mouse model of bladder cancer cells[13], suggesting that such an effect (i.e. DNMT3 inhibition) may also occur in honeybees. Therefore, inhibition of the Apis mellifera DNMT3 activity and/or expression by one or more of royal jelly’s polyphenols would regulate the expression of key genes for larval development.
Table 1. Molecular docking of 13 polyphenolic compounds of royal jelly with the Apis mellifera DNMT3 protein.
Product PubChem ID mol MW XP GScore MMGBSA dG Bind (kcal/mol) 1 Luteolin-7-O-glucoside 5280637 448.382 −10.39 −52.8 2 Luteolin-4-O-glucoside 12304737 448.382 −10.27 −47.9 3 Kaempferol 3-O-glucoside 5282102 448.382 −8.9 −64.85 4 Isorhamnetin 5281654 316.267 −7.42 −43.55 5 Hesperetin 72281 302.283 −7.42 −43.55 6 Quercetin 5280343 302.24 −7.1 −43.56 7 Pinobanksin 73202 272.257 −5.9 −37.06 8 Sakuranetin 73571 286.284 −5.76 −40.08 9 Chrysin 5281607 254.242 −5.7 −42.8 10 Naringenin 439246 272.257 −5.7 −41.05 11 Coumestrol 5281707 268.225 −5.68 −41.93 12 Genistein 5280961 270.241 −5.28 −35.7 13 Acacetin 5280442 284.268 −5.11 −37.58 Molecular docking, MMGBSA analysis, and MD simulation were carried out to identify the lead candidate polyphenolic compounds from royal jelly that can inhibit the DNMT3 protein. The binding affinity of 13 polyphenolic compounds in royal jelly for the Apis mellifera DNMT3 was evaluated using two basic metrics, XP GScore and MMGBSA dG Bind. The highest binding affinity was observed for luteolin-7-O-glucoside with a docking score of −10.3 and kaempferol-3-O-glucoside with −8.9. Furthermore, luteolin-7-O-glucoside and kaempferol-3-O-glucoside showed high total binding energies of −52.8 and −64.85 kJ/mol, respectively. MD simulations show that luteolin-7-O-glucoside maintains a consistent interaction with the DNMT3 protein throughout the simulation period. The compound luteolin-7-O-glucoside stands out as the most promising candidate and is likely to be the polyphenolic component of royal jelly responsible for most of the Apis mellifera DNMT3 inhibitory activity in this diet.
-
Homology modeling was conducted using the SWISS-MODEL server[14]. The sequences of DNA methyltransferase 3 [Apis mellifera] (ID: ADH84015.1) were obtained from NCBI. The quality of the generated models was evaluated using SAVESv6.1 − Structure Validation Server (
https://saves.mbi.ucla.edu/ ).Molecular docking
-
The DNMT3 protein structure and compounds were prepared for the docking process by atom bonds assignment, the addition of hydrogen, and energy minimization. The active site was then determined using SiteMap module (Schrodinger suite). Extra Precision docking protocol of Maestro (Schrödinger, LLC, New York, NY, 2020) was used to study the possible interaction between compounds and proteins.
MMGBSA analysis
-
The MMGBSA dG Bind (Molecular Mechanics Generalized Born Surface Area) is another method used to predict the free energy of binding of ligands to their target proteins. It provides a more refined estimate of binding free energies by incorporating solvation effects and entropic contributions. The MMGBSA module of Maestro (Schrödinger, LLC, New York, NY, 2020) was used for the calculations, with poses generated from XP docking used as input.
Molecular dynamic simulations
-
Molecular dynamic simulation was used to study the stability of the best compounds with DNMT3, using the Maestro Desmond module, 50 ns run time. More details on MD simulation methods as in previous published work[15].
-
The binding affinity of 13 polyphenolic compounds in royal jelly for the Apis mellifera DNMT3 was evaluated using two basic metrics, the docking score XP GScore and MMGBSA dG Bind to find the most promising compounds acting as inhibitors of the DNMT3 protein in honeybees. More negative values of both XP GScore and MMGBSA dG Bind indicates higher binding affinity[16]. The XP GScore values generated from the DNMT3 protein docking ranged from −10.39 to −4.2 (Table 1). With an XP GScore of −10.39, compound luteolin-7-O-glucoside has the highest binding affinity to DNMT3, indicating that compound luteolin-7-O-glucoside has a much higher binding affinity to DNMT3 than the other compounds investigated (Table 1). The MMGBSA dG Bind values varied from −64.85 to −23.33 kcal/mol, demonstrating a wide range of binding affinities. Luteolin-7-O-glucoside has ranked second among the 13 polyphenolic compounds in royal jelly in terms of the binding free energy (MMGBSA dG Bind of −52.8 kcal/mol) with DNMT3, consistent with its highest XP GScore of −10.39 (Table 1). Figure 1 shows the 3D interaction of DNMT3 with luteolin-4-O-glucoside during the induced fit docking process. MD simulations reveal the interactions of DNMT3 with luteolin-7-O-glucoside throughout 50 ns, providing insight into the binding dynamics and stability of the complex (Fig. 2). The RMSD plot provides a clear representation of the root mean square deviation (RMSD) for both the ligand (luteolin-7-O-glucoside) and the protein backbone (DNMT3) throughout the simulation (Fig. 2). The blue line, representing the protein backbone, initially shows fluctuations but gradually stabilizes around the 30 ns mark, suggesting that the protein conformation reaches a relatively stable state. Similarly, the red line, corresponding to the ligand, shows fluctuations indicative of interaction dynamics and eventually stabilizes in tandem with the protein backbone. This stabilization indicates that the luteolin-7-O-glucoside maintains a consistent interaction with the DNMT3 protein throughout the simulation period (Fig. 2).
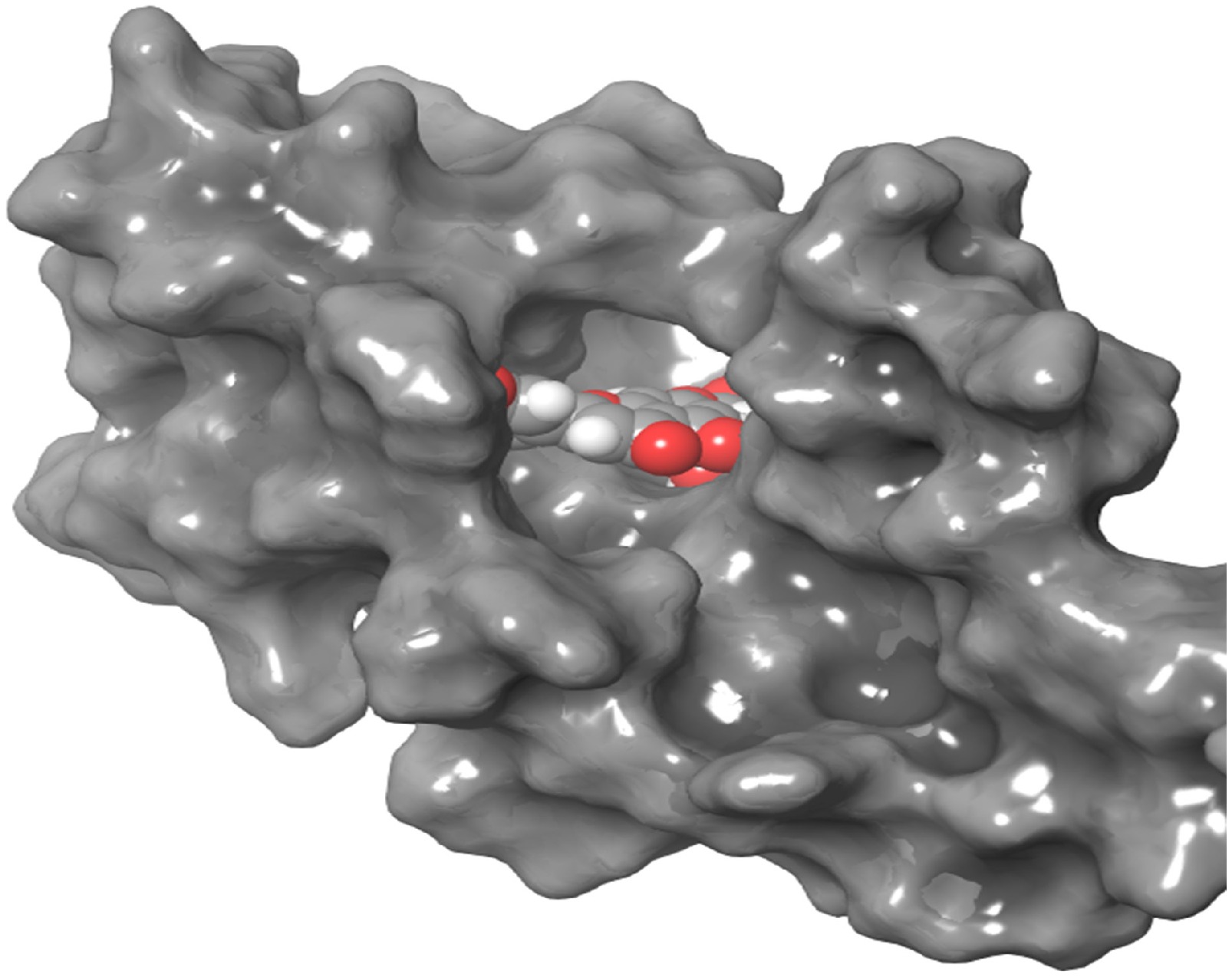
Figure 1.
3D interaction of DNMT3 with luteolin-4-O-glucoside. The compound is shown in the center with balls in different colors.
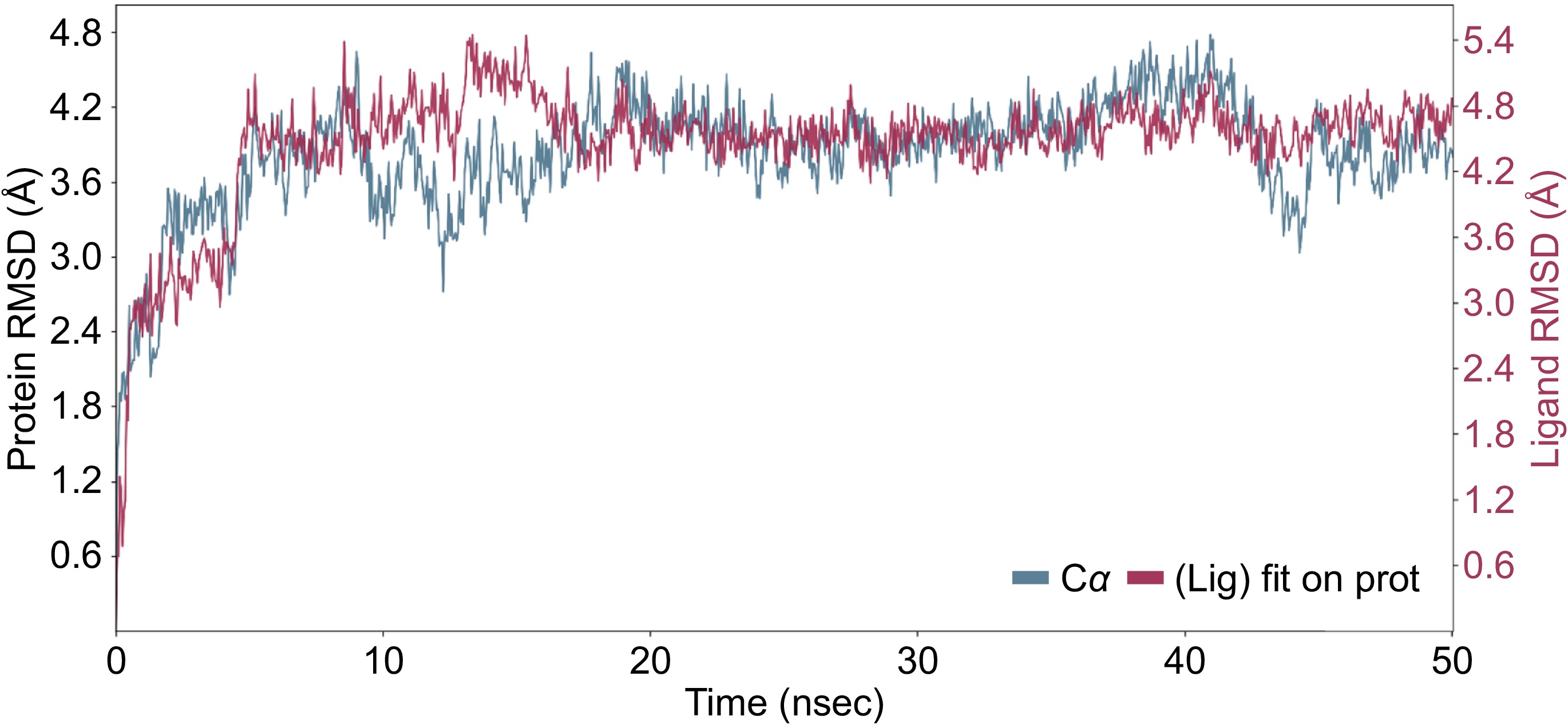
Figure 2.
MD simulation result of the interaction of DNMT3 and luteolin-4-O-glucoside during 50 ns simulation period. The RMSD showing the interaction of the ligand (red) with the protein backbone (blue).
The interaction histogram provides a quantitative summary of the percentage of interactions between luteolin-7-O-glucoside and the different residues of the DNMT3 protein (Fig. 3a). Key residues such as TYR 11, ILE 13, GLU 15, PHE 30, ASP 86, PHE 90, and TYR 93 show high interaction percentages, indicating their important role in DNMT3 binding to luteolin-7-O-glucoside. The histogram categorizes these interactions, with color coding to distinguish types such as hydrogen bonds and hydrophobic interactions. Green bars indicate hydrogen bonds, purple bars hydrophobic interactions and blue water bridges. This visual representation highlights the importance of specific residues in maintaining the binding affinity of luteolin-7-O-glucoside to DNMT3 (Fig. 3a).
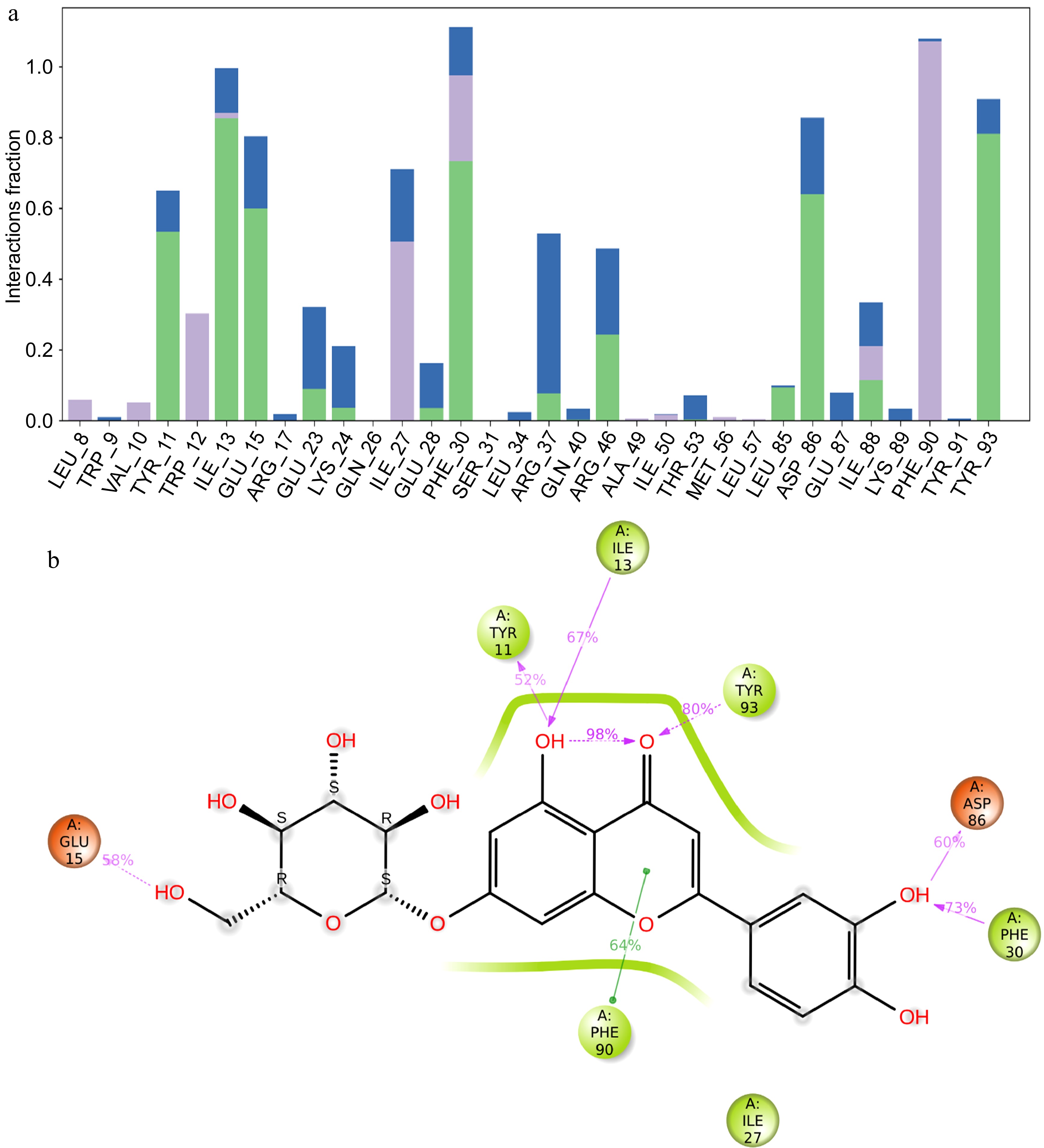
Figure 3.
MD simulation result of the interaction of DNMT3 and luteolin-4-O-glucoside. (a) Histogram showing the percentage of interacted residues with the compound, green bars indicate hydrogen bonds, purple bars hydrophobic interactions and blue water bridges. (b) 2D interaction of luteolin-4-O-glucoside with DNMT3 protein residues, residues colored according to charge, hydrogen bonds in violet and hydrophobic bonds in green.
The 2D interaction diagram further illustrates the specific interactions between DNMT3 residues and luteolin-7-O-glucoside (Fig. 3b). Residues are color-coded according to their charge properties, providing a clear visual distinction. Hydrogen bonds, shown in violet, highlight significant binding interactions with residues such as TYR 11, TYR 93, ASP 86, and GLU 15 of the DNMT3 protein, emphasizing their essential role in binding to luteolin-7-O-glucoside. As shown in Fig. 2c, TYR11 forms a hydrogen bond 52% of the time, ILE13 67%, TYR93 80%, ASP 86 60%, GLU15 58%, and PHE30 73% of the time. Hydrophobic interactions, shown in green, reveal interactions with residues such as PHE 90, indicating areas where the ligand interacts with non-polar regions of the DNMT3 protein (Fig. 3b). This detailed diagram provides a comprehensive view of the binding interface, showing the different types of interactions that stabilize the ligand-protein complex. They highlight the stability of the complex and the specific residues involved in maintaining this stability, providing valuable insights into the molecular interactions involved.
Analysis of the interaction between DNMT3 and the compound kaempferol-3-O-glucoside
-
In the present study, the compound kaempferol-3-O-glucoside has ranked second in terms of binding affinity with an XP GScore = −8.9 and first in terms of the binding free energy (MMGBSA dG Bind = −64.85 kcal/mol) (Table 1). Figure 4 shows detailed interactions between DNMT3 and the kaempferol-3-O-glucoside compound during the simulation period. Initially, from 0 to 10 ns, the RMSD gradually increases, indicating that the protein is undergoing some structural adjustments as it stabilizes. Between 10 and 30 ns, the RMSD values stabilize around 3−4 Å, indicating that the protein has reached a relatively stable conformation. In the final phase, from 30 to 50 ns, there are slight fluctuations around 4−5 Å, showing that the protein retains some conformational flexibility even after stabilization. For the ligand backbones, residues remained with the protein backbones, indicating the stability of the ligand within the binding pocket of the protein (Fig. 4).
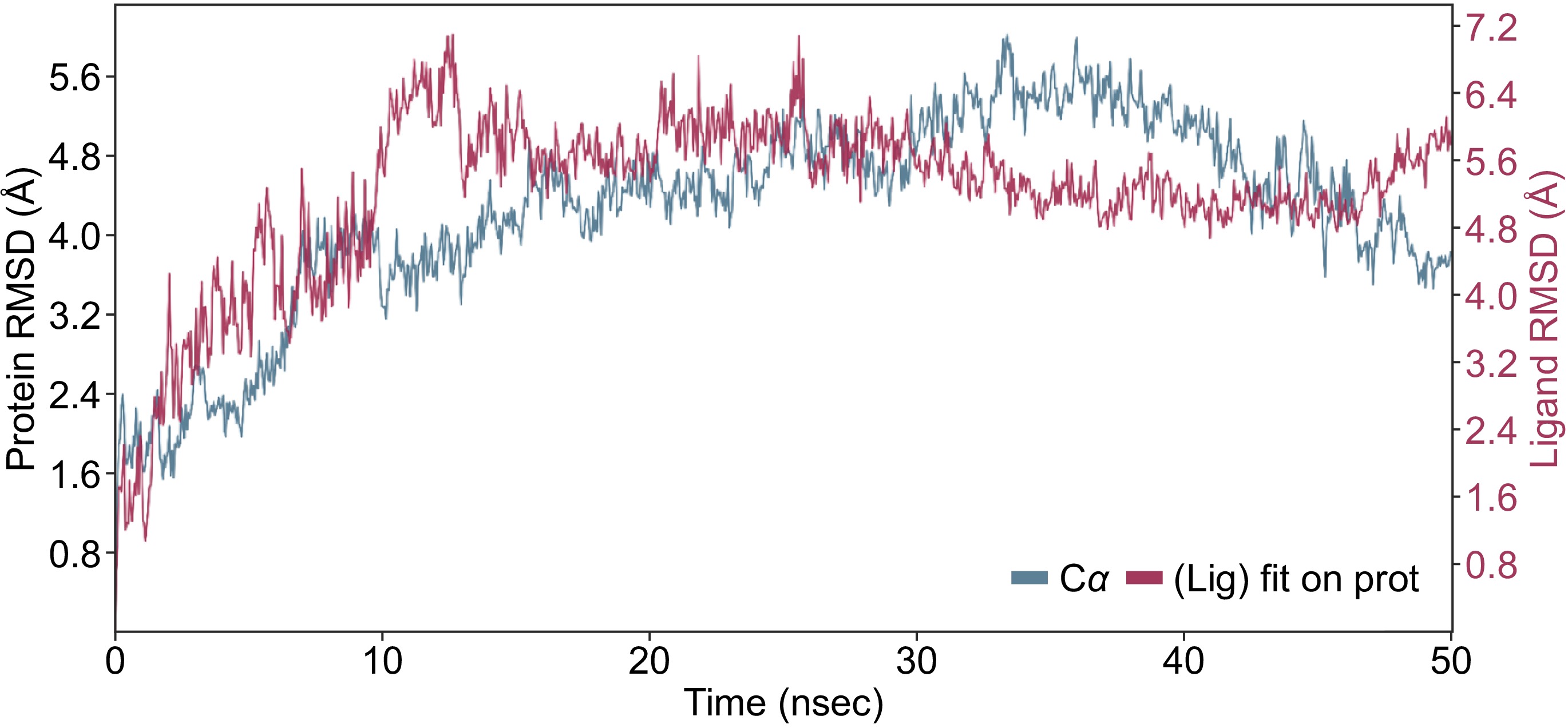
Figure 4.
MD simulation result of the interaction of DNMT3 and kaempferol-3-O-glucoside during 50 ns simulation period. The RMSD showing the interaction of the ligand (red) with the protein backbone (blue).
The histogram in Fig. 5a provides further details on the types of interactions that occur between the DNMT3 protein and kaempferol-3-O-glucoside. Key residues such as TRP12, ALA49, and ILE50 show significant hydrophobic interactions. Residues such as TYR 11, ILE 13. ARG37 and TYR 93 are predominantly involved in hydrogen bonding. This diversity of interactions highlights the complex nature of the binding affinity between DNMT3 and kaempferol-3-O-glucoside. Figure 5b details the percentage of residues associated with ligand binding during the simulation period. For example, TYR11 forms a hydrogen bond 76% of the time, ILE13 52%, ARG37 60%, and TYR93 56% of the time. These hydrogen bonds are crucial for the stability and specificity of the ligand binding. In addition, residues such as ILE50, ALA49, and TRP12 are involved in significant hydrophobic interactions that contribute to the overall stability of the ligand within the binding pocket.
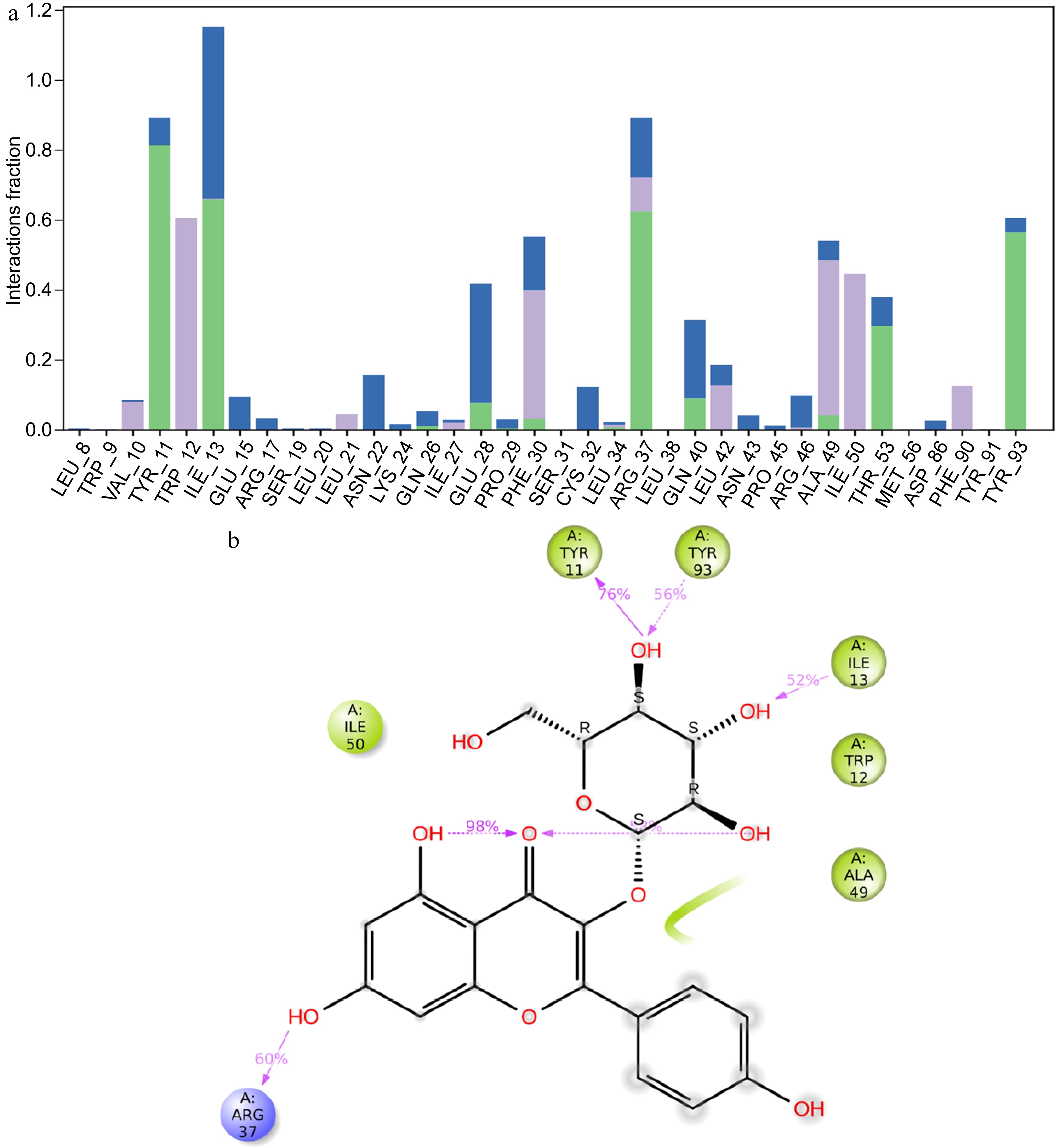
Figure 5.
MD simulation result of the interaction of DNMT3 and kaempferol-3-O-glucoside. (a) Histogram showing the percentage of interacted residues with the compound, green bars indicate hydrogen bonds, purple bars hydrophobic interactions, and blue water bridges. (b) 2D interaction of luteolin-4-O-glucoside with DNMT3 protein residues, residues colored according to charge, hydrogen bonds in violet and hydrophobic bonds in green
-
Hemi-methylated DNA produced during DNA replication is specifically targeted by DNMT1 to maintain genomic methylation, while DNMT3A and DNMT3B methylate the cytosine of unmethylated CpG sites on both DNA strands to perform de novo DNA methylation[17,18]. DNMT3-mediated DNA methylation is required for development[17,19−22] and is also essential for phenotypic changes in adult female bees in response to nutritional input (i.e. royal jelly)[8].
Experimentally, inhibiting DNMT3 has provided important clues to understanding its physiological and pathophysiological roles. When DNMT3 is inhibited with siRNA in larvae, 72% of adult bees become queens with fully developed ovaries identical to those of queens reared on pure royal jelly in the hive[8], suggesting that DNMT3 inhibition mimics the effect of royal jelly on caste phenotype. The present study aimed to identify the lead candidate polyphenolic compounds from royal jelly that can inhibit the Apis mellifera DNMT3 protein. The two basic metrics, XP GScore and MMGBSA dG Bind, were used to assess binding affinity. Of the13 polyphenolic compounds in royal jelly docked to DNMT3 protein, the compounds luteolin-7-O-glucoside and kaempferol-3-O-glucoside appear to be promising candidates for inhibition of DNMT3 activity.
The differential feeding with royal jelly for genetically identical larvae generated two distinct female castes, fertile queens and sterile workers[1−5,7]. Interestingly, silencing DNMT3 expression in newly emerged larvae had a royal jelly-like effect on larval development, with most DNMT3-depleted individuals emerging as queens with fully developed ovaries[8]. These observations are an indication that royal jelly has biologically active compounds that specifically inhibit DNMT3.
Royal jelly contains small amounts of polyphenols (Table 1), ranging from 14 to 18,936 μg/kg[1]. Of the 13 polyphenolic compounds in royal jelly docked to the Apis mellifera DNMT3, luteolin-7-O-glucoside and kaempferol-3-O-glucoside were the highest in terms of binding affinity and total binding energy (Table 1), indicating that the two compounds could be promising inhibitors of the DNMT3 protein. In support of this, luteolin was shown to decrease the expression of DNMT3A and DNMT3B proteins in human colon cancer cells[11], and in Hela cells[12].
A major target of DNMT3-mediated DNA methylation in honeybees is the dynactin p62 gene[8,23]. The larvae fed royal jelly for long periods showed reduced activity and expression of DNMT3, together with reduced overall methylation of dynactin p62[23]. Interestingly, as a result of dynactin p62-related downstream molecular events, all emerging adults were queens, suggesting an important role for DNMT3-mediated dynactin p62 methylation in larval development. This also suggests that one or more epigenetically active polyphenols in royal jelly modulate dynactin p62 methylation. In support of this idea, luteolin has been shown to target the dynactin p62 gene in several experimental models[24−26].
The present study showed that kaempferol-3-O-glucoside is also a promising candidate for inhibition of the Apis mellifera DNMT3. Supporting this conclusion, kaempferol was shown to specifically inhibit and degrade DNMT3B protein in mouse model of bladder cancer without affecting DNMT3A or DNMT1 expression, suggesting that kaempferol is a specific inhibitor of DNMT3B[13]. Interestingly, the specific inhibition of DNMT3B by kaempferol resulted in the modulation of DNA methylation at specific regions[13]. Considering that DNMT3A and DNMT3B have different preferences for flanking sequences of CpG target sites[27−29], the selective inactivation of Apis mellifera DNMT3B by kaempferol may result in different DNA methylation patterns, further enhancing the effects of luteolin in establishing an epigenetic state necessary for larval development into a queen.
Binding efficiency and inhibition increased with increasing the number of hydrogen bonds formed between the ligand and the target protein[30]. Luteolin 7-O-glucoside formed six hydrogen bonds with residues namely TYR 11, ILE 13, GLU 15, PHE 30, ASP 86, and TYR 93 (Fig. 3b), whereas kaempferol 3-O-glucoside formed four hydrogen bonds with residues TYR 11, ILE 13. ARG37, and TYR 93 (Fig. 5b; Table 2).
Table 2. Interactions and binding energies of luteolin-4-O-glucoside and kaempferol-3-O-glucoside with the Apis mellifera DNMT3.
Product Structure No. of hydrogen bonds XP GScore MMGBSA dG Bind (kcal/mol) Hydrogen bond interactions Luteolin-7-O-glucoside 
6 −10.39 −52.8 TYR 11, ILE 13, GLU 15, PHE 30,
ASP 86, and TYR 93Kaempferol 3-O-glucoside 
4 −8.9 −64.85 TYR 11, ILE 13. ARG37, and TYR 93 The honeybee genome encodes only one DNMT3 protein, consisting of 758 amino acids, whose catalytic domains have a high similarity to human DNMT3A and human DNMT3B, reaching 61% and 66% respectively[19]. The present study showed that luteolin-7-O-glucoside and kaempferol-3-O-glucoside bind to several residues located in the N-terminal domain of the Apis mellifera DNMT3, which contains the DNA-binding domain[19]. The binding of the polyphenolic compounds to the N-terminal domain of the DNMT3 could lead to a decrease in its DNA methyltransferase activity. This conclusion is supported by the fact that the DNA-binding activity of the N-terminal domain of human DNMT3A contributes to the DNA methyltransferase activity of this enzyme[31,32]. Interestingly, human DNMT3A showed high DNA binding and DNA methylation activities, while no such activities were observed with the other isoform, DNMT3A2, which is also encoded by the DNMT3A gene but lacks the N-terminal 219 amino acid residues[31].
Luteolin-7-O-glucoside and kaempferol-3-O-glucoside showed the highest binding affinity and the highest total binding energies among the 13 polyphenolic compounds in royal jelly docked to the DNMT3 protein. The compound luteolin-7-O-glucoside appears to be the most promising candidate for inhibiting DNMT3 activity in honeybees. This could be attributed to 1) its highest docking score (XP GScore −10.39), 2) the increase in its hydrogen bonding with DNMT3 (six bonds), 3) the maintenance of a consistent interaction with the DNMT3 protein throughout the simulation period, and 4) a high binding free energy, second only to kaempferol-3-O-glucoside (MMGBSA dG Bind = −52.8 kcal/mol).
-
The production of queens with fully developed ovaries when DNMT3 is inhibited in the larvae provides strong evidence that royal jelly contains epigenetically active compounds that act as inhibitors of DNMT3 to create and maintain the epigenetic state necessary in the developing larvae to produce a fertile queen. To date, the epigenetically active compounds in royal jelly with inhibitory effects on DNMT3 are unknown. The present study was designed to identify the lead candidate polyphenolic compounds from royal jelly that can inhibit the DNMT3 protein. Thirteen polyphenolic compounds in royal jelly were docked to the Apis mellifera DNMT3 protein. Of the 13 compounds docked, the top two compounds with high binding energies were luteolin-7-O-glucoside and kaempferol-3-O-glucoside. Luteolin-7-O-glucoside stands out as the most promising candidate and is likely to be the polyphenolic component of royal jelly responsible for most of the DNMT3 inhibitory activity in this diet, thereby determining developmental fate. To confirm these predictions, the effects of a special diet consisting of worker jelly rich in luteolin-7-O-glucoside on the development of larvae into adult bees need to be studied to elucidate whether such a diet rich in luteolin-7-O-glucoside mimics the effect of royal jelly in terms of DNMT3-related phenotypic changes that occur in adult female bees.
Royal jelly is widely used as a dietary supplement in alternative medicine for the treatment of various conditions, including infertility. Some animal studies have shown that royal jelly may affect the female reproductive system[33,34]. It will therefore be of great interest to evaluate whether the well-documented role of royal jelly in the development of larvae into queens and the beneficial effects of this diet on the reproductive system in female animals also applies to humans, particularly in the context of drug treatment of female infertility.
-
Not applicable.
-
The author confirms sole responsibility for the following: study conception and design, data collection, analysis and interpretation of results, and manuscript preparation.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
-
The author declares that there is no conflict of interest.
- © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Alhosin M. 2024. Luteolin-7-O-glucoside and kaempferol 3-O-glucoside are candidate inhibitors of the Apis mellifera DNMT3 protein. Epigenetics Insights 17: e001 doi: 10.48130/epi-0024-0001 

Luteolin-7-O-glucoside and kaempferol 3-O-glucoside are candidate inhibitors of the Apis mellifera DNMT3 protein
- Received: 19 August 2024
- Revised: 01 September 2024
- Accepted: 04 September 2024
- Published online: 19 September 2024
Abstract: Honeybees use royal jelly-controlled DNMT3-mediated epigenetic mechanisms to produce two distinct female castes, a long-lived fertile queen and a short-lived sterile worker. DNMT3 inhibition in larvae mimics the effect of royal jelly in terms of phenotypic changes that occur in adult female bees. A key question to be addressed in the honeybee genome is to identify epigenetically active compounds in royal jelly that inhibit DNMT3 and thereby determine developmental fate. Molecular docking, MMGBSA analysis, and MD simulation were performed to identify the lead candidate polyphenolic compounds from royal jelly that inhibit DNMT3. Thirteen polyphenolic compounds were docked to DNMT3 and two basic metrics, XP GScore and MMGBSA dG Bind, were used to evaluate the binding affinity. The highest binding affinity was observed for luteolin 7-O-glucoside with a docking score of −10.3 and kaempferol 3-O-glucoside with −8.9. Furthermore, the two compounds exhibited high total binding energies of −52.8 and −64.85 kJ/mol, respectively. MD simulations show that, unlike kaempferol 3-O-glucoside, luteolin-7-O-glucoside maintains a consistent interaction with the DNMT3 throughout the simulation period. These results suggest that of the 13 polyphenolic compounds in royal jelly, luteolin-7-O-glucoside is the most promising candidate to be the component responsible for most of the DNMT3 inhibitory activity in this diet.
-
Key words:
- Honeybees /
- Epigenetic /
- Royal jelly /
- DNA methyltransferase 3 /
- Polyphenols.