









-
Genetic variations and epigenetics play important roles in regulating flavonoid accumulation in plants. Genetic variation refers to DNA sequence differences present in the genomes of different individuals, impacting coding sequences, promoter regions, and regulatory elements. These differences can directly or indirectly influence gene expression levels and regulatory patterns, thereby impacting the plant's phenotype. The most common genetic variation is single nucleotide polymorphism (SNP), resulting from changes in a single nucleotide in the DNA sequence. They can be classified as deletion, insertion, or substitution mutations. Research has indicated that SNPs in coding regions can alter amino acid sequences, impacting protein function. Numerous studies on the diversity of gene sequences related to flavonoid biosynthesis have been conducted in plants such as Arabidopsis thaliana[1], maize[2], mango[3], barley[4], and tomato[5]. The results revealed a significant correlation between SNP content in genes associated with flavonoid biosynthesis and the accumulation of flavonoid compounds. Furthermore, genetic variations lead to phenotypic differences among cultivars of important crops like jujube[6], camellia[7], hemp[8], bamboo[9], rice[10], and maize[11]. Plant phenotypic changes are influenced not only by genetic variations but also by epigenetic factors[12]. Epigenetics refers to heritable changes in gene expression patterns without altering the DNA sequence, encompassing DNA methylation, histone modifications, and chromatin remodeling[13]. DNA methylation is a conserved epigenetic marker across eukaryotes, and it is involved in many important biological processes, such as human aging[14], cancer development[15], genome integrity[16], gene imprinting, and plant stress responses. Studies have shown that DNA methylation impacts the accumulation of flavonoid compounds in plants. For instance, in Lithocarpus polystachyus Rehd[17], Triticum aestivum L.[18] and tomatoes[19], DNA methylation occurring in promoter and coding regions regulates gene expression associated with flavonoid biosynthesis, impacting flavonoid compound accumulation. DNA methylation plays a pivotal role in crucial biological processes in seeds[20], embryos[21], and flowers[22]. Consequently, both genetic and epigenetic variations significantly contribute to flavonoid compound accumulation in plants.
However, sequence variations and epigenetic modifications are not independent phenomena, they interact intricately and culminate in phenotypic transformation. Methylated cytosines are more prone to induce a higher mutation rate at the sequence level, leading to changes in methylation levels and consequently influencing gene expression. Previous studies have shown that in Arabidopsis[23], lotus[24], and sugarcane[25], methylated cytosines are more susceptible to mutation compared to unmethylated cytosines, resulting in CG=>TG type variations and causing changes in methylation that impact plant gene expression. However, SNPs can also lead to changes in the sequence type or methylation levels of DNA. When cytosine is mutated to other types of bases (A, T, and G), the methylation pattern or level can also change, resulting in a difference in methylation levels at these sites[26]. Similarly, when other bases (A, T, and G) mutate to cytosine, the methylation type and methylation level were acquired, resulting in differences between the two sites. In addition, non-cytosine mutations also result in changes in methylation patterns and methylation levels, for example, CHG=>CHH and CHH=>CG. Studies in Arabidopsis[27] and cassava[26] have revealed that SNPs can change both methylation type and level. This indicated a close relationship between genetic variations and epigenetic during plant growth and development. However, current research on the co-regulatory mechanisms of genetic and epigenetic variations in plant phenotypes remains relatively limited.
Honeysuckle (Lonicera japonica Thunb.), belongs to the genus Lonicera of the family Caprifoliaceae is named for its flower development process (Fig. 1a), in which the color changes from silver-white to golden-yellow. Honeysuckle is rich in medicinally active compounds, including luteoloside, chlorogenic acid, flavonoids, and sesquiterpenes. Pharmacopeia records indicate that the flowering stage, known as the white bud (WB) stage, has the highest content of medicinally active compounds[28]. Among these, flavonoids play a crucial role in antiviral activity[29] and have been used to treat various viruses. Sijihua and Beihua No. 1 (Beihua) are the two most common honeysuckle cultivars in China and show significant phenotypic differences. Sijihua has a short WB stage, lasting only 2−3 d, whereas Beihua can have a WB stage lasting up to 20 d, allowing for a longer storage period of high-content medicinal active compounds. Additionally, the flowers exhibit conspicuous color variations at distinct developmental stages. Research has demonstrated that Beihua consistently displays higher levels of total flavonoids and other essential medicinal compounds compared to Sijihua[30]. A recent study revealed that DNA methylation in Sijihua affects the expression of carotenoid-related genes, leading to variations in flower color during different stages of development[31]. Honeysuckle, a crucial antiviral medicinal plant source, exhibits significant phenotypic differences among cultivars, including variations in metabolite accumulation and other traits. Growth and development, particularly flowering, are regulated by DNA methylation. This makes the honeysuckle an ideal material for studying the co-regulatory mechanisms of genetic variations and epigenetics in flavonoid compound synthesis.
The release of the honeysuckle genome has laid the foundation for studying the genomic structure of different cultivars[32]. Whole-genome resequencing technology to sequence honeysuckle will provide a comprehensive and high-resolution view of genetic variations among different cultivars. We aimed to elucidate the molecular mechanisms underlying phenotypic differences and accumulation of active compounds in different honeysuckle cultivars from the perspectives of genetic and epigenetic variations. In this study, multi-omics approaches were employed, including whole-genome resequencing, whole-genome bisulfite sequencing, and transcriptome sequencing. This comprehensive investigation focused on Beihua No. 1 (Beihua) and Sijihua during two key stages (WB and GF), characterized by differences in phenotypes and metabolite accumulation. Using whole-genome resequencing, a large number of genetic variations were identified a pseudo-reference genome was constructed for Beihua, single-base resolution DNA methylation profiles were compared between Beihua and Sijihua at different flowering stages, the characteristics of DNA methylation changes examined in different contexts. The relationship between SNPs and differentially methylated cytosines (DMCs) in honeysuckle genomes during two developmental stages were explored. Furthermore, the impact of SNPs on key enzyme-encoding genes involved in the flavonoid biosynthesis pathwaywere analyzed, thereby revealing crucial factors contributing to the differences in flavonoid biosynthesis and accumulation between the two cultivars. This study reveals the molecular mechanisms by which genetic variations and epigenetic co-regulation to phenotypic differences and variations in metabolite accumulation in the two honeysuckle cultivars. This study provides new insights into honeysuckle breeding and cultivation techniques and serves as a reference for exploring regulatory mechanisms of growth and development through the application of genetic variations and epigenetics in other species.
-
In this study, flower tissues in different stages of honeysuckle cultivar 'Beihua No.1' (abbreviated as 'Beihua' throughout the entire article) were collected, including juvenile bud (JB), green bud (GB), white bud (WB), silver flower (SF), and golden flower (GF), from Zhongke Honeysuckle Planting Cooperative in Pingyi County, Shandong, China (35°31'02'' N, 117°36'55'' E). Whole-genome bisulfite sequencing (WGBS) and transcriptome sequencing were performed using flower tissues from two stages of the honeysuckle cultivar Beihua, namely, the WB and GF stages. Young leaf tissue from Beihua was used for the resequencing.
Three biological WGBS libraries were sequenced using the Hiseq X10 sequencer (Illumina, San Diego, CA, USA) as paired-end 150-bp reads. Three biological libraries were generated using the VHTS Universal V6 RNA-seq Library Prep Kit following the manufacturer's instructions (Illumina). All libraries were sequenced using the Novaseq 6000 platform (Illumina, San Diego, CA, USA), and 150 bp paired-end reads were generated.
The reference genome for the cultivar 'Sijihua' of honeysuckle was obtained from the NCBI database under accession number PRJNA794868. Additionally, transcriptome data for the bud stage (WB: SRX14408207 − SRX14408209) and the flowering stage (GF: SRX144082013 − SRX144082015) were downloaded from the same database. Whole-genome bisulfite sequencing (WGBS) data for the WB stage (SRR18684863 − SRR18684865) and GF stage (SRR18684866 − SRR18684868) were also retrieved from NCBI. It is worth noting that the collection location and environmental conditions for the cultivation of the Sijihua cultivar of honeysuckle are identical to those of Beihua (Honeysuckle Planting Cooperative in Pingyi County, Shandong, China (35°31'02'' N, 117°36'55'' E)[31].
Library construction and sequencing
-
For resequencing library construction, DNA isolation was fragmented using Bioruptor (ThermoFisher Scientific, Waltham, MA, USA), resulting in libraries with approximately 300 bp fragment sizes. Quality control of the libraries was performed using the Qubit dsDNA HS Assay Kit (ThermoFisher Scientific) and Agilent 2100 Bioanalyzer System (Agilent Technologies, Santa Clara, CA, USA). High-quality DNA libraries were then sequenced on the BGISEQ-500 platform, generating reads of 150 bp in length.
WGBS libraries were constructed and prepared using the TruSeq DNA L T kit (Illumina) as described previously. Three biological WGBS libraries were sequenced on Illumina Hiseq X10 sequencers as paired-end 150-bp reads.
Total RNA was extracted from the honeysuckle cultivar 'Beihua' petals at the WB and GF stages using the cetyltrimethylammonium bromide (CTAB) method. Three biological libraries were prepared using the VHTS Universal V6 RNA-seq Library Prep Kit following the manufacturer's instructions from Illumina. Subsequently, all libraries were sequenced using the Novaseq 6000 platform from Illumina, located in San Diego, CA, USA, producing 150 bp paired-end reads.
Analysis of whole-genome resequencing data and construction of Beihua pseudogenome
-
We used FASQC (
www.bioinformatics.babraham.ac.uk/projects/fastqc ) for quality control of the generated FASTQ files, after which low-quality reads were filtered out, including those with more than 10% N content and more than 50% of low-quality bases (< 10). The net data obtained after filtering were compared to the Sijihua reference genome using BWA software (v0.7.12). Duplicates generated by PCR amplification were labeled and removed using the Picard package (https://sourceforge.net/projects/picard/ ). The 'HaplotypeCaller' function of GATK4 (version 4.1.4.1,https://hub.docker.com/r/broadinstitute/gatk/ ) was employed to generate GVCF files. Raw variant calling sets underwent hard filtering with parameters 'QD < 2.0 || MQ < 40.0 || FS > 60.0 || SOR > 3.0 || MQRankSum < −12.5 || ReadPosRankSum < −8.0'. The VCF file generated by hard filtering included the chromosome number of SNPs, SNP positions, reference bases, mutant bases, etc. Next, bedtools maskfasta was used to align the SNP information of the VCF file in the reference genome of Sijihua and finally constructed the pseudo-genome of Beihua. The downstream analysis script add_ka_ks.pl from MCScanX was used for calculating Ka/Ks (non-synonymous/synonymous) values for each gene pair.SNP annotation was performed using SNPEff software (
https://sourceforge.net/projects/snpeff/ ) based on the constructed Beihua pseudogenome, SNPs were classified into intergenic regions, upstream, downstream, Splice-site-donor, Splice-site-acceptor, UTR-5-prime, UTR-3-prime, and SNPs encoding exons were further divided into synonymous and nonsynonymous mutations.Whole genome bisulfite sequencing data analysis
-
Raw bisulfite sequencing reads of Beihua was trimmed by Trimmomatic v0.39 to obtain clean data and aligned to the replaced Beihua pseudogenome by BSMAP v2.90. A 4% mismatch rate was allowed per 150 bp of read length and only uniquely mapped reads were retained for subsequent analysis. Next, reads aligned to unmethylated lambda DNA were used to estimate the conversion rate. Whether cytosine is methylated or not was identified mainly based on the conversion rate and binomial distribution. The methylation level of each cytosine was calculated using the methration.py script of the BSMAP software. The formula for calculating the level of cytosine methylation is #C/(#C+#T), with #C representing the methylated cytosine and #T being the unmethylated cytosine. Similarly, whole genome bisulfite sequencing data of Sijihua were calculated by the same method. DMCs between the two cultivars of Beihua and Sijihua were identified using methylkit default parameters. In addition, the absolute methylation levels of white and golden flowers of both cultivars, under the same contexts of CG, CHG, and CHH, were to vary more than 40%, 20%, and 10%, respectively. In this paper, both the loss of CG, CHG, and CHH (CG-loss, CHG-loss, and CHH-loss) and the gain of CG, CHG, and CHH (CG-gain, CHG-gain, and CHH-gain) were defined as DMCs, and this type of DMCs is relative to the two extremes of variation from the presence to the absence and from the absence to the presence of the two cultivars.
Transcriptome sequencing data analysis
-
For each biological replicate of the Beihua and Sijihua transcriptome data, we used Trimmomatic v0.39 for quality control and aligned to the Beihua pseudogenome and Sijihua reference genome using HISAT2 default parameters, and we kept only uniquely mapped reads to estimate expression values (Supplemental Table S1). The expression of each gene we quantified using stringTie v2.1.7. Next, DESeq2 v1.32.0 was used to identify differentially expressed genes in white buds and golden flowers of both Beihua and Sijihua cultivars and differentially expressed genes were required to satisfy Log2|FC| > 2 and FDR < 0.05.
GO enrichment analysis
-
GOATOOLS was used for gene ontology (GO) enrichment of overlapping genes of differentially expressed genes (DEGs) in white buds and golden flowers of both Beihua and Sijihua cultivars. Only GO terms with P-values less than 0.05 were retained for analysis. Heatmap, and GO enrichment plot of DEGs were made using R software version 3.5.
-
To reveal the genomic variations between Sijihua and Beihua, Beihua was resequenced (Fig. 1a), generating 3.7 Gb of high-quality clean data. Reads were mapped to Sijihua's reference genome using BWA software. Over 91% of read pairs successfully aligned at a coverage depth of 30 × (Supplemental Table S2), facilitating subsequent genetic variation analysis. 9,909,981 SNPs were identified in the Sijihua genome. The majority of these SNPs were situated in intergenic regions (72.42%), followed by intronic regions (8.4%), upstream (8.35%), and downstream (7.34%) regions (Fig. 1b, Supplemental Table S3). SNPs were classified into four groups based on their impact on genes (
https://pcingola.github.io/SnpEff/se_inputoutput/ ). 12,688 high-impact SNPs (0.13%) resulted in stop-gained, stop-loss, splice-site acceptor, splice-site donor, and start-lost mutations. 102,188 low-impact SNPs (1.03%) resulted in synonymous coding, start gained, synonymous stop, or non-synonymous start mutations. Additionally, 158,107 moderate-impact SNPs (1.6%) caused non-synonymous mutations in the coding region. The remaining 9,636,998 SNPs (97.25%) had a modifying impact on intergenic, intronic, upstream, downstream, UTR-3-prime, and UTR-5-prime regions. A total of 38,808 genes were affected by four SNP categories. These included 7,555 genes associated with high-impact SNPs, 24,967 genes associated with moderate-impact SNPs, 23,256 genes associated with low-impact SNPs, and 38,787 genes associated with modifier-type SNPs (Fig. 1c, Supplemental Table S4). High-impact SNPs may be involved in the expression of important genes with key functions[33]. Therefore, a Gene Ontology (GO) functional enrichment analysis of genes associated with high-impact SNPs were performed. Regardless of the white bud (WB) or golden flower (GF) stage, genes related to high-impact SNPs were significantly enriched in response to stimuli, response to viruses, gene silencing regulation, and RNA interference regulation (Fig. 1d).Previous studies have reported the construction of pseudo-reference genomes with incomplete genome assembly by using genetic variation substitution in phylogenetic closed cultivars, such as to obtain pseudo-reference genomes for maize Mo17 and rice Matsumae, researchers substituted SNP information for maize B73[34], and rice RZ35[35]. Since the genomes of Sijihua and Beihua are very similar (SNP ratio of 1.12%), a pseudo-reference genome was created for Beihua by replacing the SNP sites with the reference genome of Sijihua.
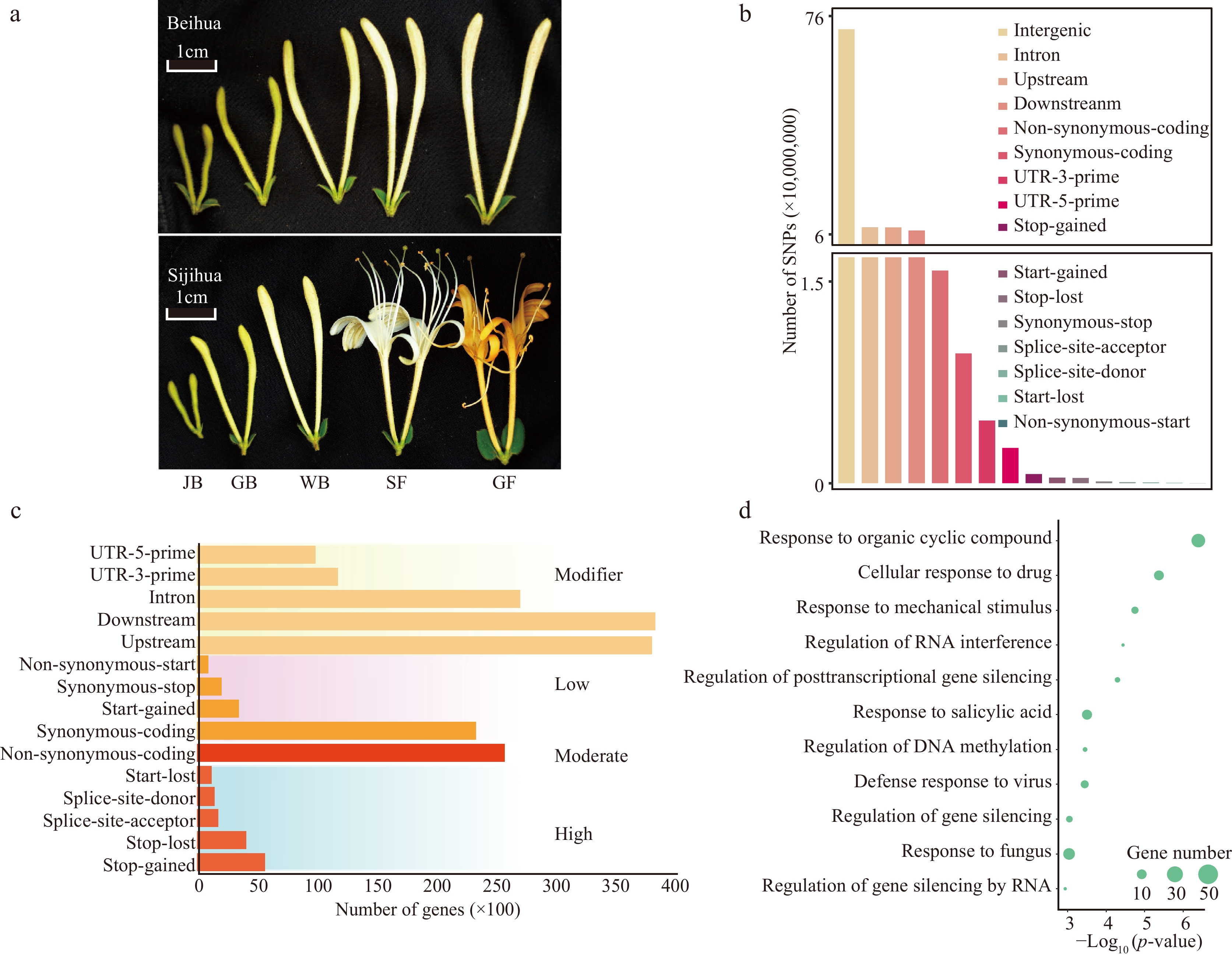
Figure 1.
Identification and classification of SNPs. (a) Different development stages of Beihua and Sijihua. Five stages are juvenile bud (JB), green bud (GB), white bud (WB), silver flower (SF), and golden flower (GF), respectively. (b) Classification of SNPs based on genome location. (c) Number of genes affected by SNPs (SNPs were divided into four impact types: high, moderate, low, and modifier). (d) GO enrichment analysis of DEGs related to high-impact SNPs.
Comparison of DNA methylation between Sijihua and Beihua
-
To investigate DNA methylation distinctions between the two honeysuckle cultivars, DNA methylation was sequenced at two stages of floral development (WB and GF) in Beihua using the WGBS technique (Supplemental Table S5). Compared with DNA methylation in Sijihua[31], no significant differences in global CG DNA methylation levels were found between the two cultivars at different developmental stages (T-test, WB: p-value = 0.17, GF: p-value = 0.62). we observed Beihua had significantly higher global DNA methylation levels than Sijihua in the CHG and CHH sequence contexts (T-test, p-value < 0.05). This phenomenon was consistently observed in both WB and GF stages (Fig. 2a). The methylation of WB and GF in Beihua and Sijihua were also analyzed. It was found that in Beihua, only the CHH methylation level showed significant differences, while in Sijihua, methylation levels in all three contexts exhibited significant differences (Supplemental Fig. S1). Furthermore, the average DNA methylation levels in 500 Kb windows across the genome were calculated and observed varying degrees of bias in all three sequence contexts. Specifically, the CG methylation distribution deviated to the right (Supplemental Fig. S2a & e), indicating that CG methylation levels were lower in Beihua than in Sijihua. The differential distribution of CHG and CHH methylation showed a significant left deviation (Supplemental Fig. S2b, f, c, & h), indicating that the methylation levels of CHG and CHH were significantly higher in Beihua than in Sijihua. These results were consistent with global DNA methylation levels (Fig. 2a).
The number of methylated cytosines were further compared between Sijihua and Beihua, revealing that Sijihua had 138,186,587 methylated cytosines, whereas Beihua had 113,582,475. During the WB stage, Sijihua had a higher number of methylated cytosines in the CG (27,577,348), CHG (21,837,724), and CHH (88,771,515) contexts compared to Beihua, which had CG (22,491,550), CHG (17,537,206), and CHH (17,537,206) methylated cytosines. A similar trend was observed during the golden flower stage, indicating that Beihua's DNA structure is less susceptible to methylation than Sijihua (Fig. 2b). When calculating the methylation levels of each cytosine, both CG and CHG methylation in the two honeysuckle cultivars showed frequent occurrences of two extremes (0 and 1), representing the completely methylated and unmethylated states, respectively. Both showed a bimodal distribution pattern. In Sijihua, 72.9% and 36.6% of the CG and CHG methylation sites, respectively, occurred in a completely methylated state, whereas in Beihua, 69% and 33.7% of the CG and CHG methylation sites, respectively, occurred in a completely methylated state. The proportion of completely methylated CG and CHG in Sijihua was higher than that in Beihua, this trend remained consistent at the GF stage (Fig. 2c & d). Furthermore, we found that the higher percentage of completely methylated CG in Sijihua, approximately 80.7% (18,525,847 / 22,956,440) of CG methylation, could be maintained from WB to GF. In contrast, in Beihua, only 76.6% (13,962,522 / 18,227,835) of CG methylation was maintained from WB to GF. This suggests that during the process of maintaining these methylation types, CG methylation in Sijihua may be more stable and faithfully replicated during DNA replication than that in Beihua[36].
To investigate methylation distribution in two honeysuckle genomes, gene density, transposable elements (TE) density, SNP density, and average methylation level were calculated in the three methylation contexts of Sijihua and Beihua (Fig. 2e). The TE-enriched regions of both Sijihua and Beihua showed higher DNA methylation levels and lower gene density at both WB and GF stages (Fig. 2e, Supplemental Fig. S2d). This phenomenon in many flowering plants, such as Arabidopsis[37], soybean[38], maize[39], rapeseed[40], and tomato[41]. SNPs showed an uneven distribution across the genome, with a high content occurring in regions with elevated CG and CHG methylation levels, while being scarce in CHH-methylated regions (Fig. 2e). To explore the relationship between DNA methylation and SNP density, the correlation between SNP density and methylation in three sequence contexts were analyzed. Significantly, we observed that SNP density was positively correlated with CG and CHG methylation levels, and negatively correlated with CHH methylation (Supplemental Fig. S3). This suggests that highly methylated cytosines may influence the stability of DNA sequences, especially the CG and CHG methylation.
To examine the methylation patterns in the coding regions of the two honeysuckles, the methylation levels within the gene coding regions and their 2 Kb flanking regions for both Sijihua and Beihua were calculated. CG methylation levels in the coding regions were higher in Sijihua than in Beihua, whereas CHG and CHH methylation levels were lower in Sijihua than in Beihua. This pattern was consistent across both WB and GF (Supplemental Figs S4a−c, S5a-c). Based on previous reports indicating the abundance of transposable element insertions in the coding regions of honeysuckles[31], the genes were classified into two types: TE-related genes and TE-unrelated genes. Subsequently, the DNA methylation patterns of these two gene types were calculated. The methylation pattern of the coding regions of TE-related genes was the same as the genome-wide methylation pattern in both honeysuckle cultivars during the two stages, whereas TE-unrelated genes showed significantly reduced methylation levels. This suggests that TE insertions play a crucial role in maintaining the DNA methylation levels in both Sijihua and Beihua (Supplemental Figs. S4d−f, S5d−f). In addition, the DNA methylation of the TE region and the 2 Kb flanking region were calculated. The CG methylation levels of TE in Beihua was higher than that in Sijihua at two stages. However, the CHG methylation level in the TE regions did not differ significantly between these two cultivars at the WB stage, but showed marked differences at the GF stage, indicating dynamic methylation levels of TEs during honeysuckle flower development. Surprisingly, CHH methylation levels of TE regions in Beihua were significantly higher than in Sijihua at both stages (Supplemental Figs S4g−i, S5g−i). Overall, the methylation levels of Sijihua and Beihua differed significantly and varied with developmental stages.
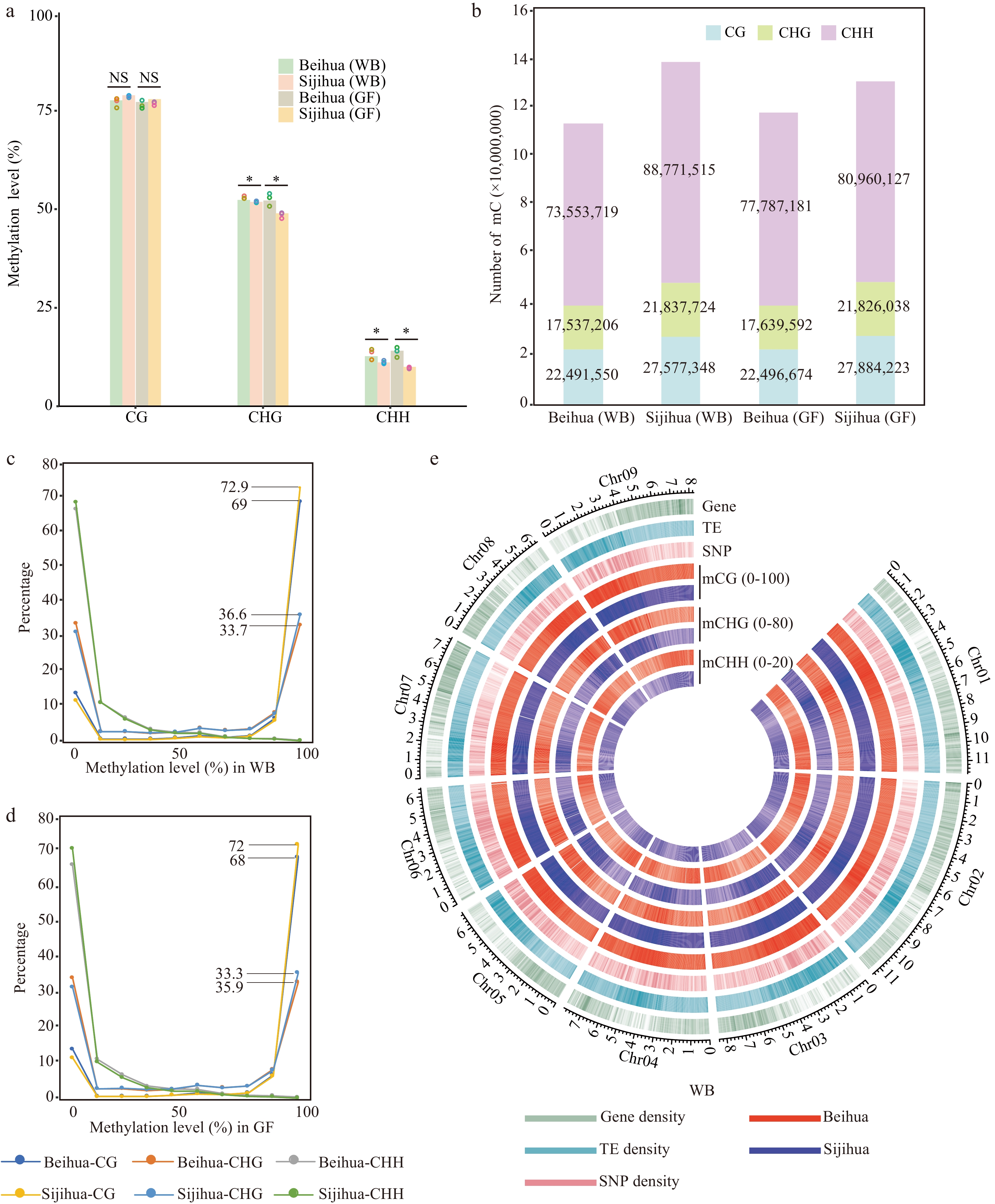
Figure 2.
DNA methylation landscape of Beihua and Sijihua. (a) Comparison of whole-genome DNA methylation levels in Beihua and Sijihua during the WB and the GF stages. Three biological replicates were calculated as dots over the bar graph (T-test, * p-value < 0.05, NS represents non-significant). (b) The relative proportions of methyl-cytosine in the contexts of CG, CHG, and CHH in Beihua and Sijihua during the WB and GF stages. (c)−(d) Distribution of methyl-cytosine methylation levels in all three sequence contexts in Beihua and Sijihua during the WB(C) and GF(D) stages were compared. (e) Circos plot showing gene density, TE density, SNP density, and DNA methylation levels for all three contexts of the WB stage.
Differentially methylated cytosines are mainly associated with SNPs
-
By comparing the DNA methylation patterns between Sijihua and Beihua, significant differences in methylation levels of all three sequence contexts were observed. To gain a deeper understanding of the specific sites displaying differential methylation, the study focused on comparing methylation levels between Sijihua and Beihua. During the WB stage, the analysis identified 1,602,266 (19%) DMCs, including 641,008 CG DMCs, 589,493 CHG DMCs, and 371,765 CHH DMCs. Notably, hyper-methylation (Sijihua > Beihua) was more prevalent than hypomethylation in three methylation contexts. This trend persisted in the GF stage (Fig. 3a). However, it is important to consider that genetic variation (SNPs) can also contribute to the variation in DNA methylation sites by influencing the sequence context or altering methylation levels[26]. To further understand how genetic variations (SNPs) impact DNA methylation sites in honeysuckles, 6,679,481 (81%) SNP-associated DMCs were identified at both WB and GF stages. It was observed that 2,539,019 DMCs resulted from cytosine loss (36%), 1,808,116 DMCs were attributed to cytosine gain (26%), and 2,332,346 DMCs were ascribed to other types of mutations (38%). For example, an illustrative snapshot from the Genome Browser displayed various SNP-associated DMCs, including CHH-gain, CG-gain, CHG-loss, CG-loss, and CHH-loss (Fig. 3b). Genetic variations can lead to changes in both methylation type and level. Moreover, it was found that the number of SNP-associated DMCs was much greater than the number of non-SNP-associated DMCs (DMCs caused by different methylation levels in the same context) when comparing differentially methylated cytosines within the two honeysuckles (Fig. 3a). These findings underscore the potential substantial contribution of genetic variations in driving disparities in DNA methylation levels between the two honeysuckles.
Regardless of the number of cytosine methylation type mutations or non-cytosine mutations, it was found that the CHH type was the most affected in terms of cytosine loss, gain, and other types of mutations, followed by the CHG and CG types (Fig. 3c & d). Among other types of mutations, the significance of guanine (G) loss and gain was particularly evident, playing a pivotal role in the dynamic transition between distinct methylation types. Transitions from CG to CHH and from CHG to CHH are the most prevalent, with a predominant proportion of guanine (G) mutations mutating to adenine (A) or thymine (T). Following this trend, mutations from CHH to CG and CHH to CHG emerged as the next most frequent occurrences, primarily involving the conversion of adenine (A) to guanine (G) (Fig. 3e). It is worth noting that these mutations not only led to changes in methylation type but also influenced the methylation levels themselves.
Specially, when CG sites underwent mutations, transforming into CHG or CHH sites, they exhibited a higher likelihood of becoming hypomethylated. Conversely, CHH sites transitioning to CHG or CG sites were more prone to hypermethylation (Supplemental Fig. S6a). Extending the examination to the diversity of the 500 bp flanking sequences surrounding the SNP-associated DMC sites, it was observed that downstream nucleotide diversity was higher than that upstream of the DMC sites. Additionally, nucleotide diversity at the DMC sites markedly exceeded that within the adjacent flanking sequences. Intriguingly, this phenomenon was not observed at non-DMC sites. The higher nucleotide diversity at DMC sites underscores their susceptibility to frequent natural selection, implying the potential for intricate interplay between SNPs and DNA methylation dynamics (Supplemental Fig. S6b).
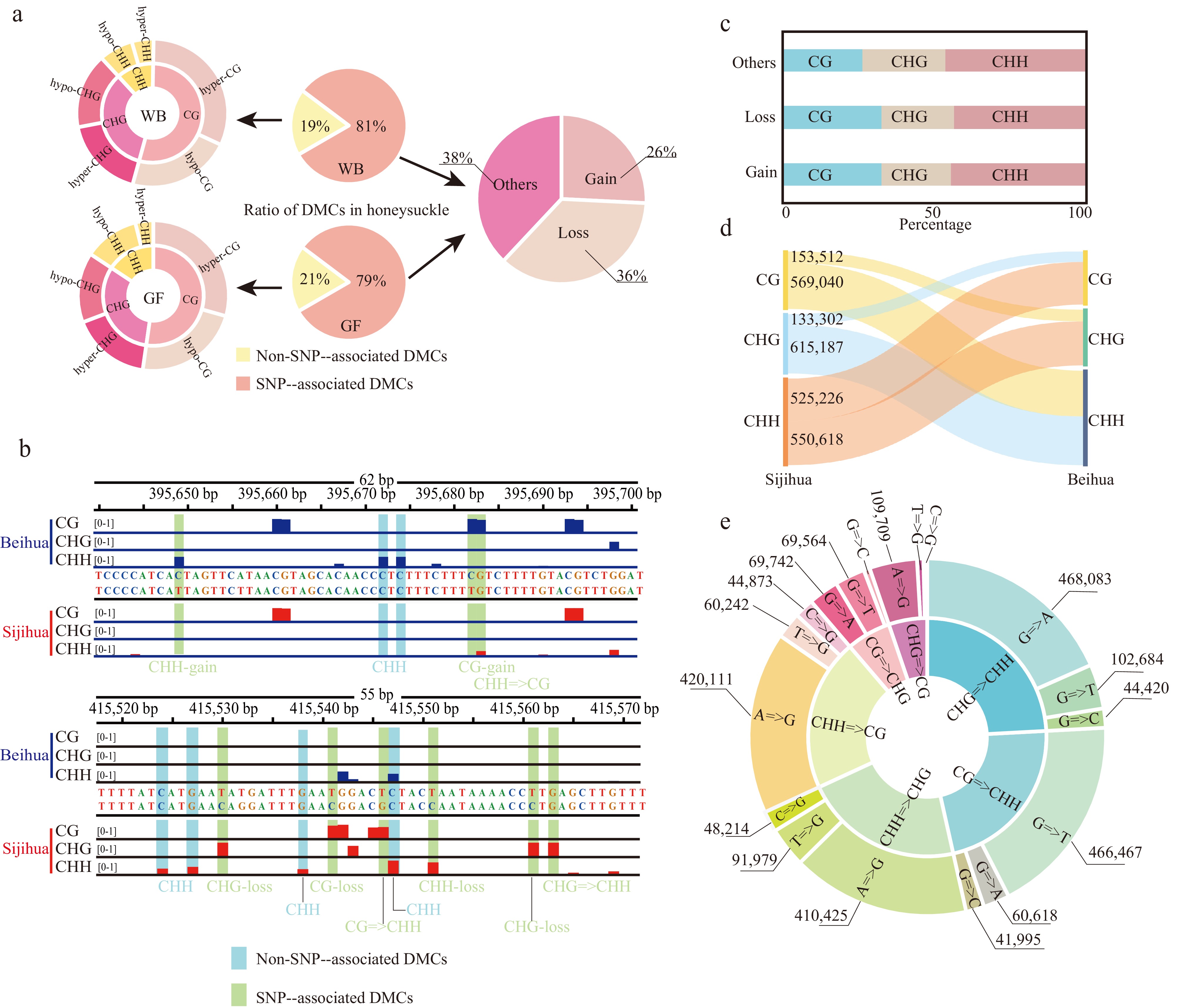
Figure 3.
Identification of differentially methylated cytosines in the honeysuckle genome. (a) The proportion of two DMC types in the honeysuckle genome, including non-SNP-associated DMCs and SNP-associated DMCs. (b) Representative screenshots of DMCs. Methylation site losses, gains, and mC context changes were indicated on the underside of the track (red: Sijihua; blue: Beihua; Light blue shading represents non-SNP-associated DMCs; Light green represents SNP-associated DMCs). (c) The proportion of methylation types of the SNP-associated DMCs. (d) Sankey diagram showing the number of changes in DNA methylation types in three contexts between Sijihua and Beihua. (e) Sunburst chart showing the number of methylation type changes caused by nucleotide mutation.
Mutations in CG=>TG affect the methylation level of genes
-
To further investigate the effect of cytosine loss on DNA methylation levels, the base substitution rates were calculated for homologous genes between Beihua and Sijihua. The rate of C=>T base substitutions was higher than that of other mutation types (Fig. 4a), consistent with studies in Arabidopsis[23] and lotus[42]. Within the honeysuckle genome, methylated cytosines exhibited a greater propensity to mutate into C=>T compared to unmethylated cytosines (1,155,283 for mC=>T, 636,736 for non-mC=>T, binomial test, p-value = 1.727e-10), resulting in a higher frequency of CG to TG mutations[24]. However, CG=>TG mutations were not evenly distributed throughout the genome. The majority of these mutations occurred in intergenic regions (272,395), followed by introns (19,132) and exons (44,634) (Fig. 4b).
To validate whether the mutation from CG methylation type to TG in honeysuckle impacted gene methylation levels, we assessed CG methylation levels within the gene body and the 2 Kb flanking regions of both Sijihua and Beihua genes. The analysis revealed significant differences in the gene body regions of both Sijihua and Beihua at both stages (Mann-Whitney test, p-value < 0.001) (Fig. 4c, Supplemental Fig. S7a). When genes unaffected by CG=>TG mutations were considered, as expected, no significant differences (Mann-Whitney test, p-value > 0.05) were observed in the gene-body regions at two stages of Sijihua and Beihua (Fig. 4d, Supplemental Fig. S7b). Subsequently, the study was focused specifically on genes with CG=>TG mutations and calculated the CG methylation levels in their gene body and 2 Kb flanking regions[43]. Intriguingly, significant differences in the gene body methylation levels between Sijihua and Beihua were observed (Mann-Whitney test, p-value < 0.001) (Fig. 4e, Supplemental Fig. S7c). This indicates that genetic variations leading to CG=>TG mutations directly affect CG methylation levels in the gene body. Furthermore, it was observed that genes affected by CG=>TG mutations not only showed reduced CG methylation levels in the gene body but also displayed a significant reduction in gene expression levels in Beihua and Sijihua (Fig. 4f−h, Supplemental Fig. S7d−f). This observation is consistent with previous findings in other plants such as Arabidopsis[44], and Brassica napus[45], where higher CG methylation within the gene body promotes gene transcription.
In summary, CG=>TG mutations not only altered the methylation levels, but also affected gene expression levels. To understand the functions of these genes affected by CG=>TG mutations, GO enrichment analysis was performed and identified their predominant enrichment in transcriptional regulation, stress response, flavonoid biosynthesis pathways, responses to viruses, biosynthesis of flavonoids, flavonols, and flavonoids (Supplemental Fig. S8). Collectively, these findings underscore the critical role played by the interactions between epigenetic and genetic variations in regulating gene functions in honeysuckle.
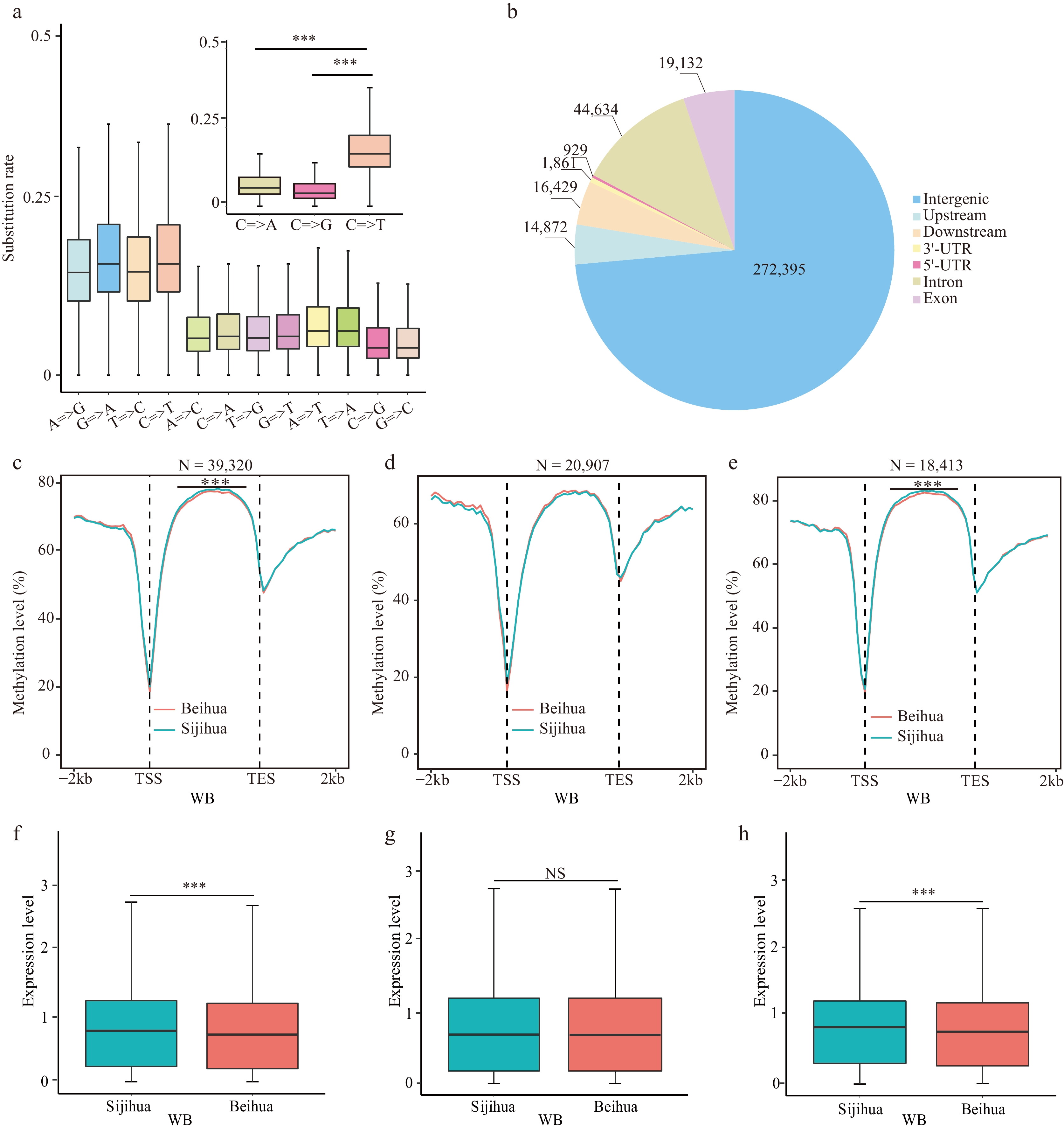
Figure 4.
Relationship of CG=>TG type mutations with CG methylation and gene expression. (a) The substitution rate of nucleotides in homologous genes between Beihua and Sijihua. (b) The distribution of CG=>TG mutation sites on the honeysuckle genome. Comparison of CG methylation levels in the body region and the flanking 2 Kb region of (c) all genes, (d) CG=>TG unrelated genes, and (e) CG=>TG-related genes during the WB stage in Sijihua and Beihua. The expression levels of (f) all genes, (g) CG=>TG unrelated genes, and (h) CG=>TG-related genes during the WB stage in Sijihua and Beihua were compared (Mann-Whitney test, *** p-value < 0.001, NS represents non-significant).
SNPs and DMCs co-regulate genes involved in the flavonoid pathway
-
To investigate deeper into the impact of SNP-associated DMCs, genes were identified that had overlaps with SNP-associated DMCs within their gene body and 2 Kb flanking regions, designating them as genes related to SNP-associated DMCs. It was found that SNP-associated DMCs were linked to 76% (29,899/39,320) of the genes within the honeysuckle genome. Among these genes, 5,324 and 7,067 genes showed differential expression (DEGs, log2|FC| > 2, FDR < 0.05) in the WB and GF stages, respectively, with an overlap of 3,325 genes displaying differential expression in both stages (Fig. 5a). Notably, the majority of genes (3,158/3,325) that exhibited differential expression at both stages showed consistent expression trends. Specifically, in both the WB and GF stages, the expression levels of these genes in Beihua were either higher than those in Sijihua (C3) or lower than those in Sijihua (C4) (Supplemental Fig. S9a). Interestingly, GO enrichment analysis of these genes with consistent expression trends revealed significant enrichment in processes related to flavonoid biosynthesis, flavonol biosynthesis, cellular glucan metabolism, stress response pathways, and floral whorl development (Supplemental Fig. S9b). Overall, genes affected by both SNPs and DMCs play important biological functions in various critical pathways, indicating their substantial regulatory significance.
The above analysis has highlighted those genes affected by both SNPs and DMCs were significantly enriched in the flavonoid biosynthesis pathway. Flavonoids represent a vital group of secondary metabolites in plants renowned for their natural medicinal properties. They play crucial roles in plant growth, development, and defense against biotic and abiotic stresses. The synthesis of flavonoids originates from phenylalanine through the phenylpropanoid pathway, involving several key enzymes (Fig. 5d)[46]. In the investigation of the effects of SNPs and DMCs on flavonoid biosynthesis in honeysuckle, 35 homologous genes encoding key enzymes in the flavonoid biosynthesis pathway in the honeysuckle genome were identified (Fig. 5d, Supplemental Table S6). Despite the presence of a high number of SNPs in both the gene body and promoter regions of these 35 homologous genes, their Ka/Ks ratios are considerably lower than 1. This suggests that these genes have undergone purifying selection during evolution and possess relatively conserved structures and sequences. Consequently, genetic variation may not be the primary driving factor behind the differences in flavonoid biosynthesis between the two honeysuckles (Fig. 5b & e).
Furthermore, by comparing the methylation levels of these 35 genes in Sijihua and Beihua at different stages, significant differences were observed in both the gene body and promoter regions. Additionally, some genes showed significant changes in expression. For example, LjPAL (EVM0007831), LjCHS (EVM0026111), LjCHI (EVM0013981), and LjFLS (EVM0027194) showed significantly higher expression levels in Beihua than in Sijihua at both the WB and GF stages (Fig. 5c, Supplemental Fig. S10). Interestingly, it was found that both the gene body and promoter regions of these 35 genes contained SNP-associated DMCs, as well as non-SNP-associated DMCs. These DMCs appear to play crucial roles in the regulation of gene expression (Supplemental Fig. S11). For example, a genome browser showed that LjCHI (EVM0013981) and LjFLS (EVM0027194) have a large number of SNP-associated DMCs and non-SNP-associated DMCs in the promoter region. Additionally, the expression levels of these genes were significantly higher in Beihua than in Sijihua (Fig. 5f & g, Supplemental Fig. S12). Therefore, the promoter regions of these key enzyme genes contain numerous nucleotide mutations that lead to changes in promoter region methylation, directly affecting gene expression. This may be a crucial factor contributing to the higher flavonoid levels in Beihua compared to Sijihua.
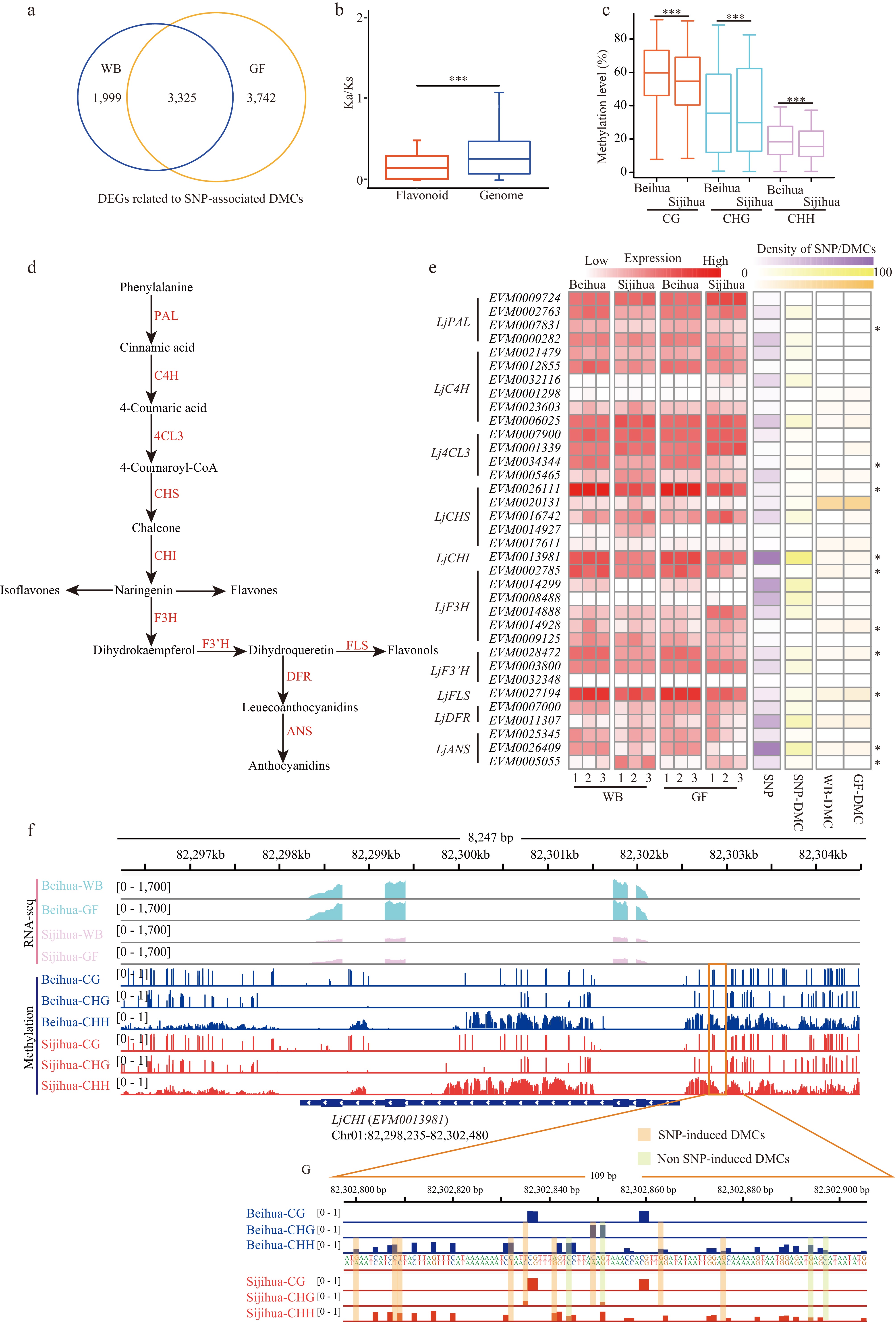
Figure 5.
The relationship between genes related to SNP-associated DMCs and the flavonoid pathway. (a) Venn diagram showing the number of overlapping differentially expressed genes (DEGs) associated with SNP-associated DMCs between WB and GF. (b) Boxplot showing Ka/Ks ratios of 35 key genes and genomes in the flavonoid pathway of honeysuckle (Mann-Whitney test. *** p-value < 0.001). (c) Boxplot showing the methylation levels of CG, CHG, and CHH in the promoter regions of 35 key genes in the flavonoid pathway during the WB in Beihua and Sijihua (Mann-Whitney test. *** p-value < 0.001). (d) The pathway of flavonoid synthesis in plants. (e) Heatmaps showing the expression levels of 35 key genes of the flavonoid pathway, the number of SNPs in the upstream region of the genes, the number of SNP-associated with DMCs, and the number of non-SNP-associated with DMCs for Beihua and Sijihua at the WB and GF (* represents DEGs in the WB and GF stages). (f) Genome browser showing DNA methylation level and expression level of LjCHI (EVM0013981). (g) Genome browser showing representative screenshots of two types of DMCs in the promoter region (Chr01: 82302800 − 82302800) of LjCHI (EVM0013981).
-
In this study, whole-genome resequencing, whole-genome bisulfite sequencing, and transcriptome sequencing techniques were employed to sequence different honeysuckle cultivars. From both the genetic and epigenetic perspectives, the factors underlying the phenotypic differences observed between the two honeysuckles were investigated. Previous studies have revealed that high-impact SNPs in the genomes of various plants, such as rice[47], tomato[48], cucumber[49], cotton[50], and peach[51], have significant effects on important agronomic traits. These high-impact SNPs have been observed to play crucial roles in plant resistance. For example, in tomato, resistance genes carrying high-impact SNPs exhibit reduced susceptibility to Oidium neolycopersici infection, resulting in enhanced resistance. In cotton, the GhDRP gene with high-impact SNPs showed decreased expression upon infection with V. dahliae, resulting in leaf yellowing, shedding, and severe cell damage. This indicates a robust lignification response to counteract pathogen attacks. The present study identified a substantial number of genes in the honeysuckle genome affected by high-impact SNPs, significantly enriched in resistance pathways, such as response to fungi, defense response to virus, and response to mechanical stimulus. These findings provide a solid foundation for improving the desirable traits of honeysuckle cultivars.
Subsequently, the genome-wide single-base resolution DNA methylomes between Beihua and Sijihua were compared. Notably, significant differences in DNA methylation were observed between these two honeysuckles in the three contexts (CG, CHG, and CHH). Some studies have suggested that changes in DNA methylation are influenced by genetic variations, subsequently regulating alterations in gene expression. In the present study, a correlation between SNP density and different methylation types was observed. Specifically, SNP density showed a significant positive correlation with CG and CHG methylation, but a negative correlation with CHH methylation. Previous reports have also indicated a positive correlation between differential methylation regions and the density of SNP variations, suggesting that genetic variations in proximity may influence some of the methylation differences observed between different cultivars[52]. Furthermore, a higher density of SNPs were also observed at sites with differentially methylated cytosines, a phenomenon previously observed in the cassava genome[26].
In the genome of honeysuckle, there is a pronounced preference for nucleotide mutations (C=>T), primarily driven by cytosine methylation. While DNA methylation plays a critical role in regulating gene expression, it can also have detrimental effects because methylated cytosines are more susceptible to deamination, leading to the conversion of cytosine to thymine. Similar phenomena have been observed in the Arabidopsis[23], canola[24], and sugarcane[25]. The interplay between genetic variation and DNA methylation has a regulatory effect on gene expression[53]. The results revealed that genes related to Beihua affected by CG=>TG had significantly lower methylation levels in the gene body compared to Sijihua, and a significant difference in the gene expression between Beihua and Sijihua.
Numerous studies have indicated that phenylalanine ammonia-lyase (PAL)[54] and chalcone isomerase (CHI)[55] served as the first and second rate-limiting enzymes in flavonoid synthesis, respectively. Overexpression of these enzymes leads to a significant accumulation of flavonoids. Additionally, the overexpression of flavanol synthase (FLS) results in a substantial accumulation of flavonoids[56]. Therefore, the promoter regions of these key enzyme genes contain numerous nucleotide mutations that lead to changes in promoter region methylation, directly affecting gene expression. This may be a crucial factor contributing to the higher flavonoid levels in Beihua compared to Sijihua. These findings align with prior research on the apple genome[57]. In summary, the interplay between SNP and DMCs in the biosynthesis of flavonoids in honeysuckle regulates the expression changes of genes related to key enzymes, thereby influencing the accumulation of flavonoids.
A large number of DMCs in the genomes of two honeysuckles were identified, with most of them being SNP-associated DMCs. These DMCs were associated with approximately 80% of the genes and were significantly enriched in several vital pathways, such as the flavonoid biosynthesis pathway, flavonol biosynthesis pathway, anticancer pathway, and cellular glucan metabolic process. In particular, SNP-associated DMCs were found in genes related to key enzymes in the flavonoid synthesis pathway. Some of these genes directly influenced the accumulation of flavonoids and exhibited significant differential expression, including LjPAL (EVM0007831), LjCHI (EVM0013981), and LjFLS (EVM0027194). Previous studies have shown that the expression levels of LjPAL, LjCHI, and LjFLS are positively correlated with flavonoid accumulation in other plants, such as Arabidopsis[55], tomato[58], Dendrobium officinale[59], and Brassica napus[60]. In summary, the interplay between genetic variation and epigenetic regulation exerts a substantial influence on gene expression, leading to noticeable phenotypic differences between the two honeysuckles. These findings provide novel insights into breeding and cultivation techniques for honeysuckles.
-
In this study, 9,909,981 SNPs were identified in Sijihua, including 12,688 high-impact SNPs that were significantly enriched in the stress resistance pathways. By comparing the DNA methylation patterns of Beihua and Sijihua, significant differences in DNA methylation levels were observed between the two honeysuckles. Thus, a substantial number of DMCs were identified between these two honeysuckles, with 81% of which were SNP-associated DMCs. Furthermore, methylated cytosines are more prone to mutation, resulting in CG=>TG, and altered DNA methylation, further regulating gene expression. SNP-associated DMCs are linked to 76% of protein-coding genes, with 3,325 genes exhibiting differential expression in both stages (WB and GF), and significantly enriched in the biosynthetic pathway of flavonoids. In the flavonoid pathway, important genes affecting flavonoid accumulation such as LjPAL, LjCHI, and LjFLS showed significant differences. The flanking 2 Kb region and body region of these genes produced a large number of SNP-associated DMCs, which is likely to be the reason that SNP-associated DMCs are regulating the overexpression of genes in Beihua, resulting in an increase in the accumulation of flavonoids in Beihua.
-
The authors confirm contribution to the paper as follows: study conception and design: Wang H; re-sequencing performing and DNA methylation analysis: Yu X; transcriptome performing and biological pathway analysis: Yu X, Yu H; tissues collection and analysis: Lu Y, Zhang C; draft manuscript preparation: Yu X, Wang H. All authors reviewed the results and approved the final version of the manuscript.
-
The raw reads generated in this study have been deposited in the CNCB sequence read archive (SRA) with the accession number PRJCA018541. The reference genome, WGBS, and RNA-seq public data of Sijihua were downloaded from the NCBI database under the project numbers: PRJNA794868, PRJNA824715, and PRJNA813701, respectively.
This work was supported by the National Natural Science Foundation of China (32160142), Guangxi Natural Science Foundation (2023GXNSFDA026034), Sugarcane Research Foundation of Guangxi University (2022GZA002), and State Key Laboratory for Conservation and Utilization of Subtropical Agro-bioresources (SKLCUSA-b202302) to Haifeng Wang.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Xianyun Yu, Hang Yu
- Supplemental Table S1
- Supplemental Table S1 Summary of Sijihua and Beihua RNA-seq reads mapping.
- Supplemental Table S2 Summary of whole genome resequencing mapping.
- Supplemental Table S3 Classification of SNPs based on genome location.
- Supplemental Table S4 Number of genes affected by SNP mutation.
- Supplemental Table S5 Summary of Sijihua and Beihua BS-seq reads mapping.
- Supplemental Table S6 Mining and analyzing flavonoid pathway genes of honeysuckle.
- Supplemental Fig. S1 The significance analysis between Beihua WB and GF, as well as Sijihua WB and GF.
- Supplemental Fig. S2 DNA methylation landscape of Beihua and Sijihua.
- Supplemental Fig. S3 The correlation between SNPs and DNA methylation.
- Supplemental Fig. S4 DNA methylation patterns of protein-coding genes and TEs in Sijihua and Beihua in the WB.
- Supplemental Fig. S5 DNA methylation patterns of protein-coding genes and TEs in Sijihua and Beihua in the GF.
- Supplemental Fig. S6 SNP lead to changes in the type and level of DNA methylation and characterization of the diversity of DMC sites.
- Supplemental Fig. S7 CG≥TG-type mutations in relation to CG methylation and gene expression during the GF stage.
- Supplemental Fig. S8 GO enrichment analysis of CG≥TG-related genes.
- Supplemental Fig. S9 The pattern of differentially expressed genes related to SNP-associated DMCs.
- Supplemental Fig. S10 The comparison of DNA methylation levels of 35 key genes of the flavonoid pathway in two stages of Sijihua and Beihua.
- Supplemental Fig. S11 Heatmaps showing the expression levels of 35 key genes, the number of SNPs in the gene body of the genes, the number of SNP-associated DMCs, and the number of non-SNP-associated DMCs in Beihua and Sijihua during the WB and GF stages (* represents genes that showed significant differences in both stages).
- Supplemental Fig. S12 Genome browser showing DNA methylation and expression levels of LjFLS (EVM0027194) and representative screenshots of two types of DMCs in the promoter region (Chr01:50581502-50581602).
- © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yu X, Yu H, Lu Y, Zhang C, Wang H. 2024. Genetic and epigenetic variations underlying flavonoid divergence in Beihua and Sijihua honeysuckles. Epigenetics Insights 17: e002 doi: 10.48130/epi-0024-0002 

Genetic and epigenetic variations underlying flavonoid divergence in Beihua and Sijihua honeysuckles
- Received: 30 July 2024
- Revised: 02 September 2024
- Accepted: 24 September 2024
- Published online: 11 October 2024
Abstract: Flavonoids are important antibacterial and antiviral active substances which are the most crucial medicinal components of honeysuckle. However, the content of medicinally active substances in different honeysuckle cultivars is significantly different. Genetic variations and epigenetics play essential roles in plant evolution and trait improvement. Here, we performed multi-omics sequencing of two honeysuckle cultivars (Beihua and Sijihua) at different stages (WB and GF). The results revealed 9,909,981 SNPs in the genomes of the two cultivars, and 12,688 high-impact SNPs were found to regulate genes involved in important biological pathways, such as plant stress resistance. Furthermore, it was found that the majority of differentially methylated cytosines (DMCs, 81%) between Beihua and Sijihua were associated with SNPs. SNP-related DMCs were associated with 76% of the genes, among which 3,325 DEGs (e.g., LjPAL, LjCHI, and LjFLS) were significantly enriched in the flavonoid biosynthesis pathway. The presence of a large number of SNP-related DMCs in the flanking and gene regions of these genes may have led to the overexpression of the genes in Beihua, which increased the accumulation of flavonoids in Beihua. In summary, the present study provides theoretical and technical support for improving the genetic and epigenetic traits of honeysuckle.
-
Key words:
- Lonicera japonica /
- Genetic variations /
- Epigenetics /
- DNA methylation /
- Flavonoid