









-
Epigenetics refers to heritable changes in gene expression that do not alter the DNA sequence but affect gene activity, such as DNA methylation, histone modifications, and non-coding RNAs. In plants, the dicer-like (DCL) and argonaute (AGO) gene families not only participate in non-coding RNA production and function but also in RNA-directed DNA methylation (RdDM)[1−3]. RNA interference (RNAi) is a critical biological process that involves both post-transcriptional gene silencing (PTGS) and transcriptional gene silencing (TGS) mediated by small RNAs. This process begins with the generation of small RNAs, which are then incorporated into the RNA-induced silencing complex (RISC). The generation of mature small RNAs (sRNAs) is primarily facilitated by DCL proteins, whereas the AGO proteins play a pivotal role as carriers, guiding sRNAs to recognize and base-pair with target mRNA sequences, ultimately regulating gene expression[2]. Beyond transcriptional and post-transcriptional silencing, DNA methylation represents another critical regulatory mechanism in various plant growth and developmental processes[1,3]. RdDM, often referred to as the canonical RdDM pathway, is a widespread epigenetic regulatory mechanism in plants. Both canonical and non-canonical RdDM pathways heavily rely on the functions of DCLs and AGOs[4].
DCLs function as molecular factories for processing plants small RNAs (sRNAs), serving highly conserved roles across plant biology. These proteins typically contain several domains, including DExD, Helicase-C, DUF283, PAZ, RNase III, and dsRNA-binding, all belonging to the ribonuclease III family[5]. The Arabidopsis thaliana (A. thaliana) genome contains four DCL genes, designated as DCL1 through DCL4, each playing a unique role in RNA silencing and plant physiological responses. For example, DCL1 is primarily responsible for the biogenesis of microRNAs, indirectly affecting normal plant development and environmental adaptation[6]. DCL2 mainly produces small interfering RNAs (siRNAs), which are crucial for plant defense mechanisms and developmental processes[7−9]. DCL3 is predominantly involved in the synthesis of 24-nucleotide siRNAs and is essential for the RdDM pathway, maintenance of genomic stability, regulation of gene expression, and responses to environmental stimuli[10,11]. DCL4 produces 21-nucleotide siRNAs, which play key roles in post-transcriptional gene silencing, especially in antiviral defense mechanisms[12]. Additionally, DCL5 (previously known as DCL3b) is found in monocots; it enhances the activity of DCL3 and plays a specialized role in reproductive processes[13].
In plants, the AGO family genes interact with sRNAs to form RISC, which act as specific regulators of gene expression across various biological processes. AGO proteins modulate gene expression through several mechanisms including transcript cleavage, suppression of PTGS, and influencing DNA methylation through RdDM, along with other specialized functions[14,15]. The AGO family exhibits significant evolutionary diversity and can be categorized into three main phylogenetic groups: AGO1/5/10, AGO2/3/7, and AGO4/6/8/9[16,17]. AGO1 is a widely expressed member that plays a central role in multiple sRNA-mediated silencing pathways, especially those associated with PTGS[18,19]. The function of AGO5 is less well understood but it is thought to be involved in gene silencing during viral infections[20,21]. AGO10 selectively binds to 21-nt siRNAs and is involved in transcriptional gene silencing pathways[22,23]. AGO2 is recognized for its role in defense against viruses, it also binds to 21-nt siRNAs to participate in PTGS[24−26]. AGO3 and AGO7, despite being phylogenetically close to AGO2, display functional divergence—AGO3 binds to 24-nt siRNAs and primarily participates in RdDM to maintain genomic and transposon stability[27], whereas AGO7 interacts with miR390 to trigger the production of trans-acting siRNAs from TAS3 transcripts[28]. AGO4, a core component of the RdDM pathway, guides 24-nt siRNAs to DNA sites to promote DNA methylation, thereby silencing their target genes[1]. AGO6 shares functional similarities with AGO4, often acting as its functional complement. Additionally, AGO9 has been shown to also participate in RdDM[29−31]. The specific roles of AGO8 and its associated siRNAs remain unclear. However, AGO8, along with its paralogs AGO4, AGO6, and AGO9, is crucial for early megaspore formation[30,32].
Although extensive research has detailed the evolution and function of DCLs and AGOs in plants, most studies have focused on their classification and diversity, with less attention given to their potential functional divergence during evolution[33,34]. In the present study, the distribution, evolution, and expansion of DCLs and AGOs were examined across a wide range of species. By constructing phylogenetic trees, their possible evolutionary trajectories within angiosperms were inferred. Their expression profiles were further analyzed in various tissues and under different stress conditions to explore the potential functions of these two gene families. The findings significantly advance the understanding of the functional evolution of DCLs and AGOs in angiosperms, and offer valuable insights that could inform future breeding strategies aimed at developing improved plant varieties.
-
To investigate the origins and evolutionary histories of DCL and AGO genes in plants, this study used Arabidopsis DCL and AGO genes as seed sequences. BLAST software was employed to identify homologous sequences in 36 plant species, spanning groups such as rhodophytes, chlorophytes, charophytes, bryophytes, ferns, gymnosperms, basal angiosperms, monocots, and eudicots. Additional validation with InterProScan confirmed the presence of requisite domains in the identified sequences. This screening process resulted in the identification of 113 DCLs and 334 AGOs across the 36 species.
To delineate the evolutionary relationships among the DCL and AGO genes, phylogenetic trees were constructed using the maximum likelihood method. The phylogenetic analysis of DCLs revealed two main branches, which can be further divided into four clades (Fig. 1a). This tree suggests that the DCL genes originated in rhodophytes and remained relatively stable in chlorophytes and charophytes. Notably, a significant expansion of DCLs occurred in bryophytes, marked by the emergence of the DCL1, DCL3, and DCL4 clades, with seed plants exhibiting widespread representation across all four clades. The AGO gene family tree consists of three main branches and seven clades (Fig. 1b), with phylogenetic evidence indicating that the ancestors of the AGO4/6/8/9 and AGO2/3/7 groups were present in algae. These genes underwent further expansion in bryophytes and ferns and fully evolved in seed plants. Taken together, these results highlight a largely consistent evolutionary history for DCLs and AGOs, suggesting synchronous evolution among these gene families. Additionally, the identification of DCL and AGO members were expanded using transcriptome-based gene annotations from the 1KP database, which covers over 1,000 plant species (Supplementary Figs S1 & S2). The results from this broader analysis are consistent with those derived from the initial 36 species. Based on sequence homology and phylogenetic insights, the origins and evolutionary trajectories of the DCL and AGO gene families across various plant lineages have been inferred, providing a comprehensive overview of their development through evolutionary history.
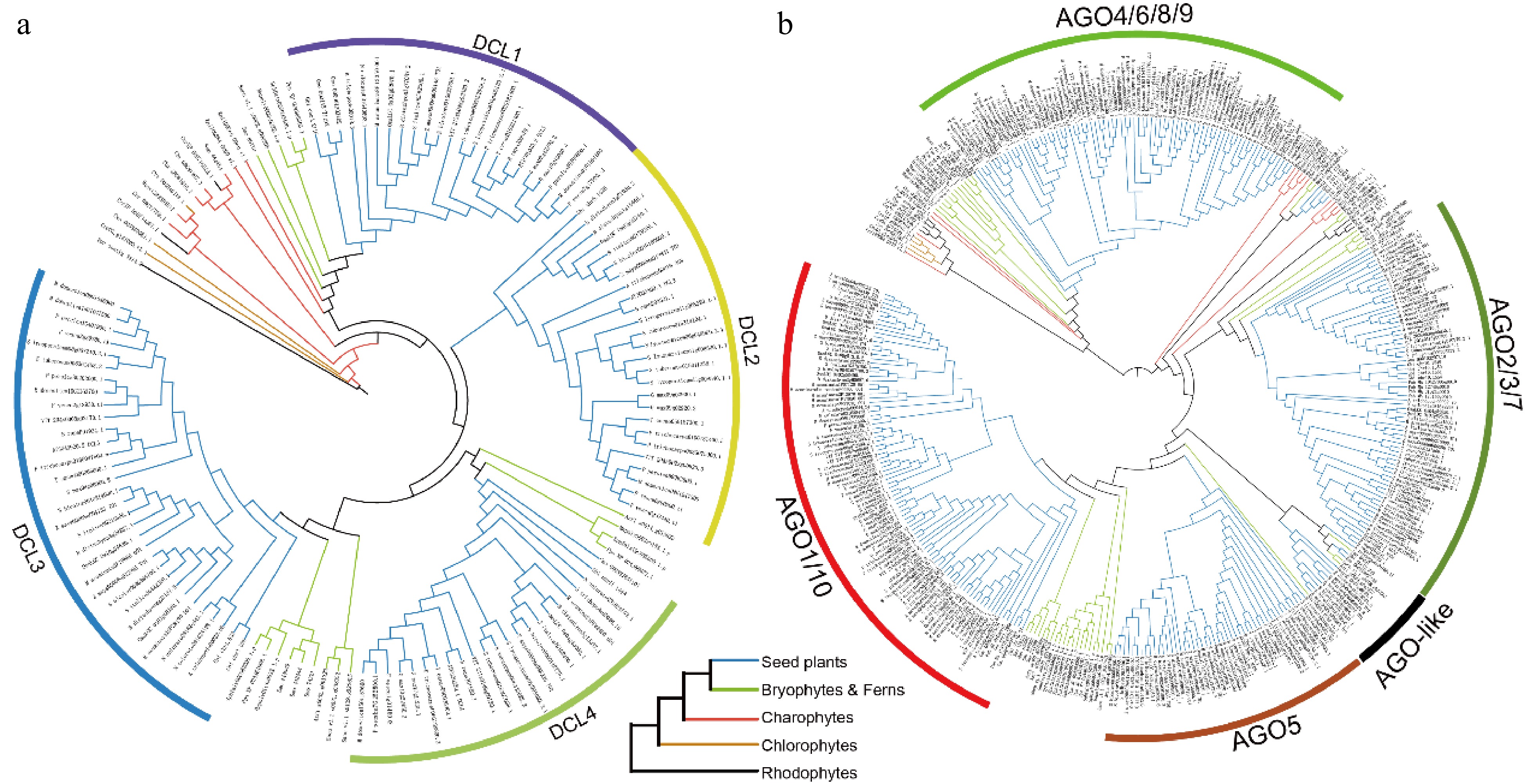
Figure 1.
Phylogenetic trees of the (a) DCL, and (b) AGO gene families across 36 plant species. Branches are color-coded to denote different plant groups: black for rhodophytes, yellow for chlorophytes, red for charophytes, green for ferns and bryophytes, and blue for seed plants.
Multiple sequence alignments of sRNA-related functional domains in DCLs and AGOs were performed. Overall, the RNase III domains of DCLs in all four branches are highly conserved at both the C- and N-termini. Additionally, there are variations among the RNase III domains between different branches, with those in higher plants being more conserved (Supplementary Fig. S3). This conservation may reflect adaptations to diverse environmental pressures through more sophisticated RNA regulatory mechanisms, leading to the synthesis of a wider variety of sRNAs. Furthermore, we aligned the MID and PAZ domains of AGOs across different branches. Members of the AGO2/3/7 and AGO4/6/8/9 branches have largely lost the MID domain, and those that retain it show less conservation. In contrast, the AGO1/5/10 branch retains a highly conserved MID domain (Supplementary Fig. S4). Similarly, the PAZ domain shows significant differences among branches, with high conservation in the AGO1/5/10 branch, including in lower plants, while the other two branches exhibit lower conservation (Supplementary Fig. S5). Highly conserved MID and PAZ domains are typically associated with fundamental miRNA processing, whereas less conserved domains may relate to specific functional requirements and adaptive changes.
In plants, the copy number of DCL genes does not appreciably vary across different evolutionary branches (Fig. 2), ranging from 25 to 40 across the four groups and predominantly existing as single-copy genes throughout plant evolution. The primary factor influencing this gene copy number variation among different plants is attributed to whole-genome duplication (WGD) events during specific evolutionary processes. By contrast, the copy number of AGO genes exhibit considerable variation across branches. Specifically, the major clades of AGO4/6/8/9, AGO2/3/7, and AGO1/5/10 contain 95, 93, and 173 genes, respectively (Fig. 2). The number of AGO genes notably exceeds that of DCL genes and shows diversification into more clades, suggesting that the AGO genes are more frequently retained during duplication events. Additionally, AGO4 and AGO6, which encode key enzymes in plant methylation through siRNA processing were analyzed separately (Fig. 2). Results show that AGO4 is prevalent in basal angiosperms, whereas AGO6 is restricted to monocots and dicots. Based on these findings, it is hypothesized that the evolution of AGO4 and AGO6 may be linked to significant shifts in reproductive strategies and the development of floral organs during the transition from gymnosperms to angiosperms. Given the unique and critical roles of AGO4 and AGO6 in methylation processes, the expansion and loss of these genes was investigated within the AGO4/6/8/9 clade across 36 species (Fig. 3). Phylogenetic analysis reveals that these genes exist as single copies in algae, ferns, gymnosperms, and basal angiosperms. Following the ε duplication event, both AGO4 and AGO6 were retained in embryophytes. Unlike AGO6, which did not undergo significant expansion after its formation, AGO4 experienced multiple duplication events. In the Brassicaceae, the α and β duplication events led to the emergence of AGO8 and AGO9. Similarly, duplication events in crops such as potatoes, tomatoes, and monocots also contributed to the expansion of AGO4.
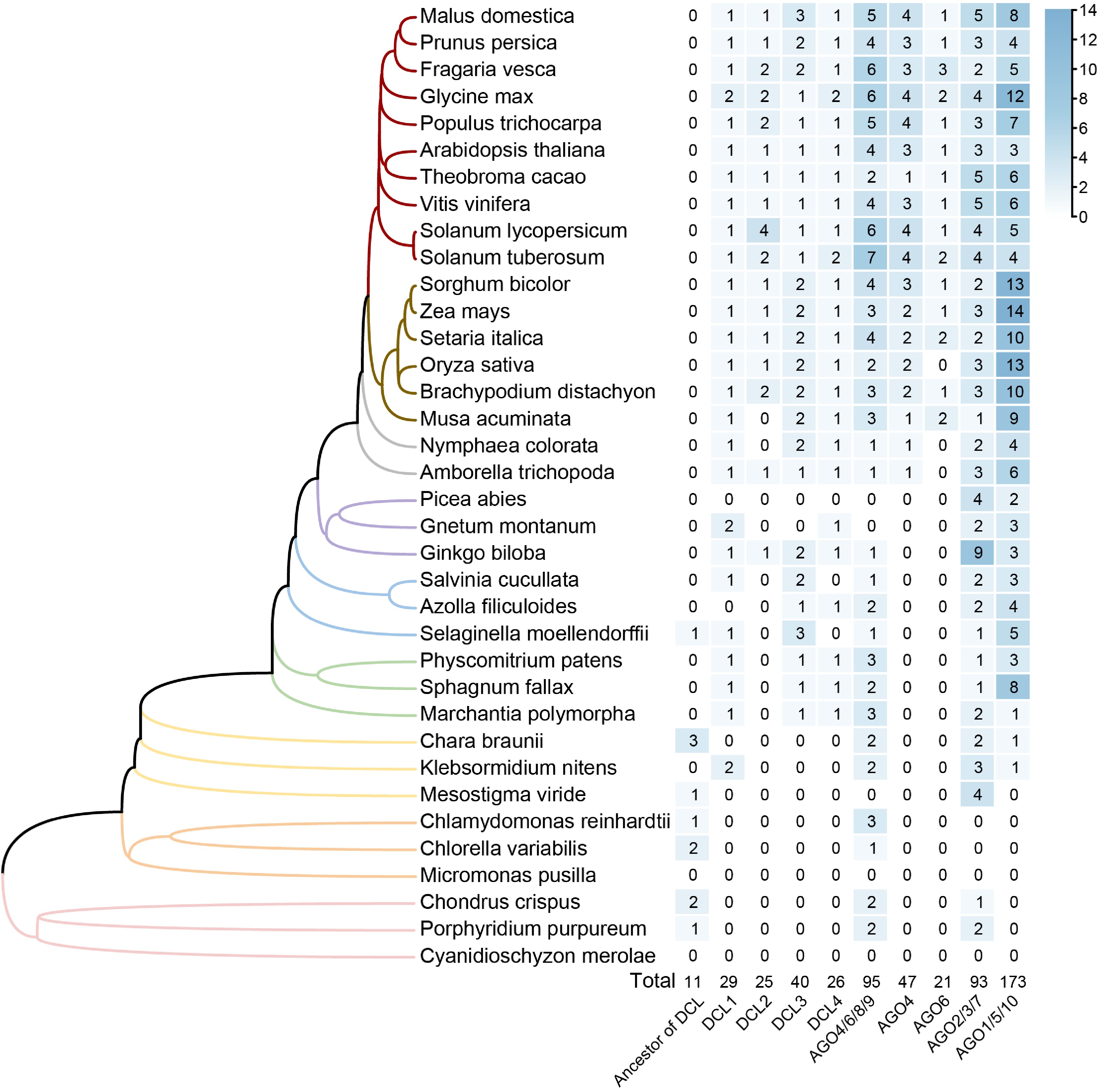
Figure 2.
Phylogenetic distribution and gene copy number analysis of DCL and AGO gene families across 36 plant species. The phylogenetic tree on the left represents the evolutionary relationships of species investigated, with branches colored to represent different groups. The heatmap on the right displays gene copy numbers for each clade of gene family across the species, with higher numbers represented by darker shades. The total counts for each clade across all species are provided at the bottom of the heatmap.
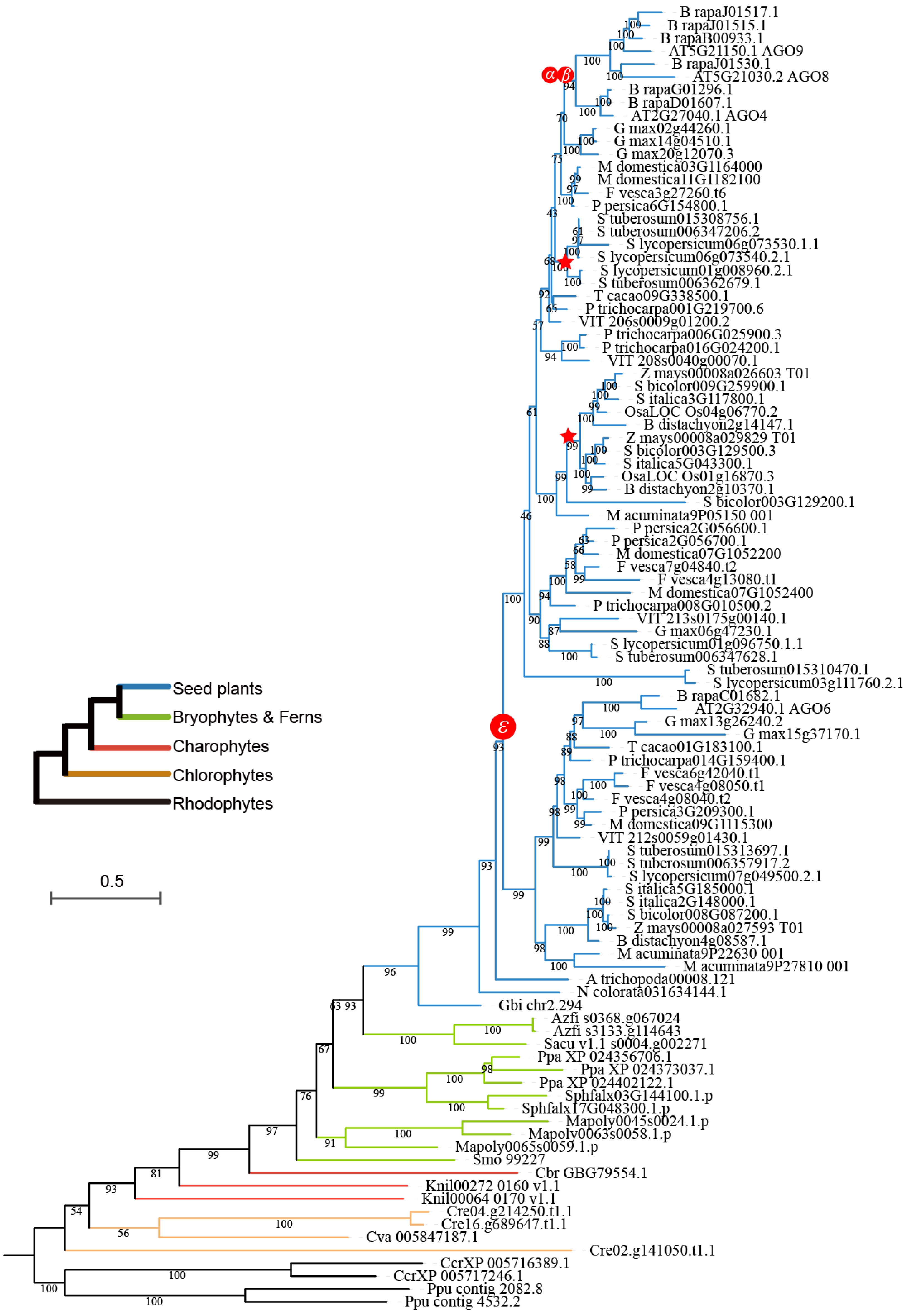
Figure 3.
Phylogeny of the AGO4/6/8/9 clade within the AGO gene family. Different colored branches represent distinct plant groups. The symbols ε, α, and β represent the epsilon angiosperm-wide WGD event, the alpha duplication event, and the beta duplication event, respectively. Red stars along the branches indicate specific whole-genome duplication events.
Phylogenetic and collinearity network analysis of the DCLs and AGOs
-
To elucidate the evolutionary differences between DCLs and AGOs in plants, a collinearity network analysis was conducted on 18 plants, including both monocots and dicots, based on their phylogenetic relationships. The analysis identified 542 syntenic gene pairs, grouping the DCL genes into four clusters that represent four distinct evolutionary trajectories. Additionally, four WGD events and three tandem duplication pairs were detected in apple, soybean, and tomato, suggesting lineage-specific expansions of the DCLs during evolution (Fig. 4a). Therefore, we integrated both phylogenetic and collinearity data were integrated (Fig. 4b) and the evolutionary history of DCL genes was reconstructed. It is proposed that two ancestral DCL genes existed before the emergence of seed plants, which subsequently underwent two WGD events. This process resulted in the loss of three branches, leaving four extant DCL groups.
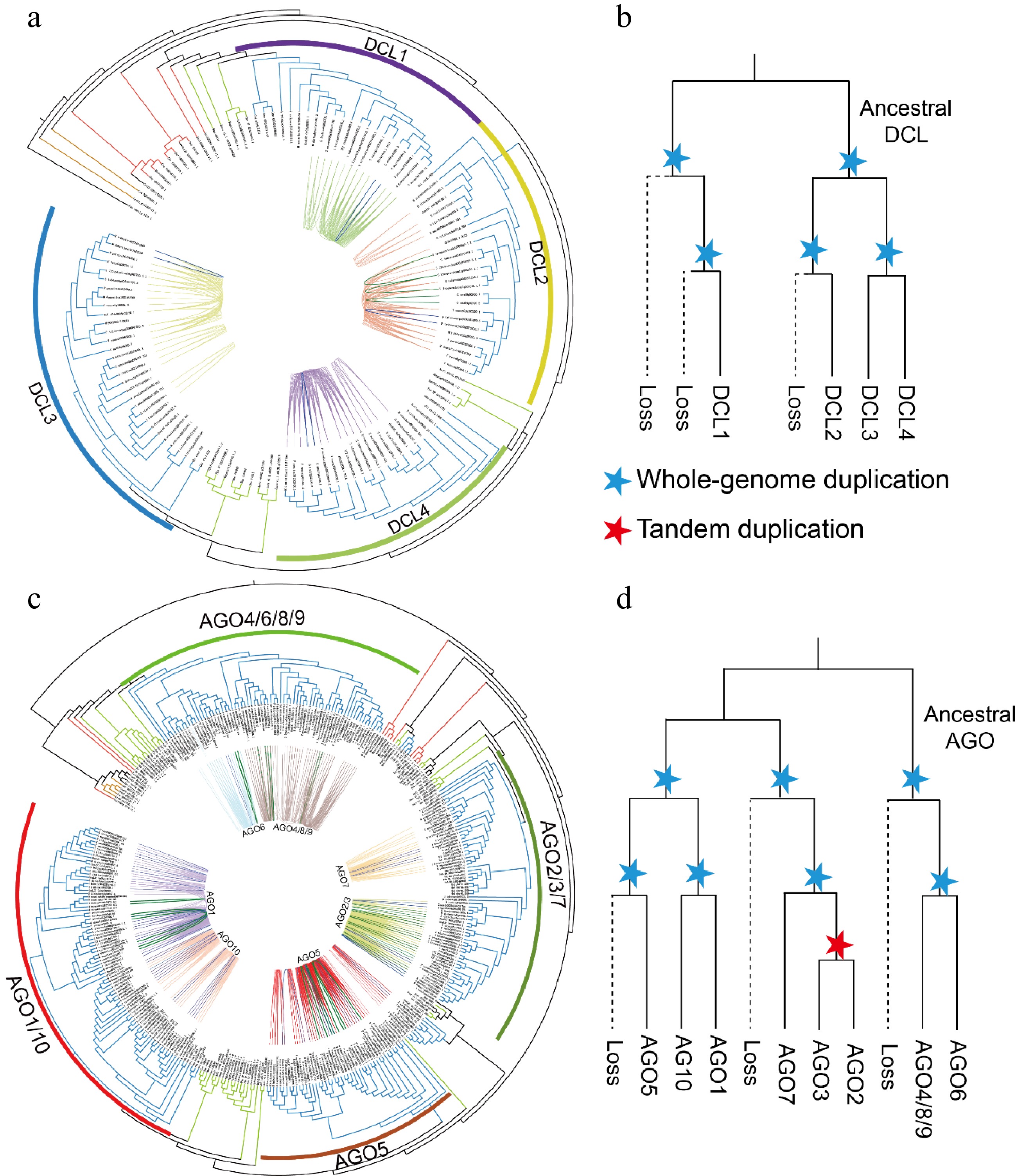
Figure 4.
Phylogenetic analysis and synteny identification of the DCL and AGO genes. (a), (c) Phylogenetic and syntenic relationships of the DCLs and AGOs. The blue and green lines indicate gene pairs resulting from WGD and tandem duplication in the DCLs and AGOs, respectively. (b, d) Schematic representation of the proposed evolutionary histories of the DCL and AGO gene families. The dashed lines indicate gene loss. Blue stars mark either the ancient seed plant-wide or angiosperm-wide genome duplication events. Red stars represent tandem duplication events of genes.
In the AGO gene family, 1,090 syntenic gene pairs were identified and subsequently clustered into seven groups (Fig. 4c). Notably, AGO4, AGO8, and AGO9 formed a cluster, demonstrating their evolutionary homology. A similar homologous relationship is observed between AGO2 and AGO3. Additionally, 43 syntenic gene pairs resulting from intraspecific duplications across various AGO groups were found. Moreover, 23 tandem duplication pairs were identified, they distributed across the syntenic gene clusters of AGO1, AGO4/8/9, AGO5, and AGO6, with most tandem duplications occurring within AGO2/3. Based on these findings, it is inferred that the AGO family originated from three ancestral genes before the emergence of seed plants, with clusters retained through two WGD events. Furthermore, AGO2 and AGO3 appear to have arisen from tandem duplications (Fig. 4d).
Expression patterns of DCLs and AGOs in key green plants
-
To explore the functional differences between DCLs and AGOs throughout plant evolution, expression patterns were analyzed using publicly available data from various tissues of P. patens, Z. may, A. thaliana, and F. vesca. Among these species, F. vesca exhibits the highest number of DCL members, totaling six. The expression profiles of various tissues were largely consistent across the four species, with DCLs showing high expression in reproductive tissues (Fig. 5a, Supplementary Fig. S6a). In F. vesca, the DCL2 and DCL3 branches each contain two gene members, however, in each branch, only one gene exhibits high expression. This contrasts with Z. mays, where the expression patterns of the two DCL3 members are similar. In P. patens, DCL3 is more prominently expressed in vegetative tissues, whereas in A. thaliana, Z. may, and F. vesca, it shows high expression in reproductive tissues. For the AGOs, the overall expression profiles are similar to those of the DCLs, with high expression in reproductive tissues (Fig. 5b, Supplementary Fig. S6b). The AGO4/6/8/9 groups demonstrate functional complementation in reproductive tissues in ancestral species, whereas in A. thaliana, Z. mays, and F. vesca, AGOs are ubiquitously expressed in all reproductive tissues. In P. patens, AGO1 group members are highly expressed only in reproductive tissues in P. patens, however, they maintain high expression levels across all tissues in A. thaliana and F. vesca. Interestingly, despite their overall low expression levels in pollen both DCLs and AGOs from various groups remain active in reproductive organs in A. thaliana and F. vesca.
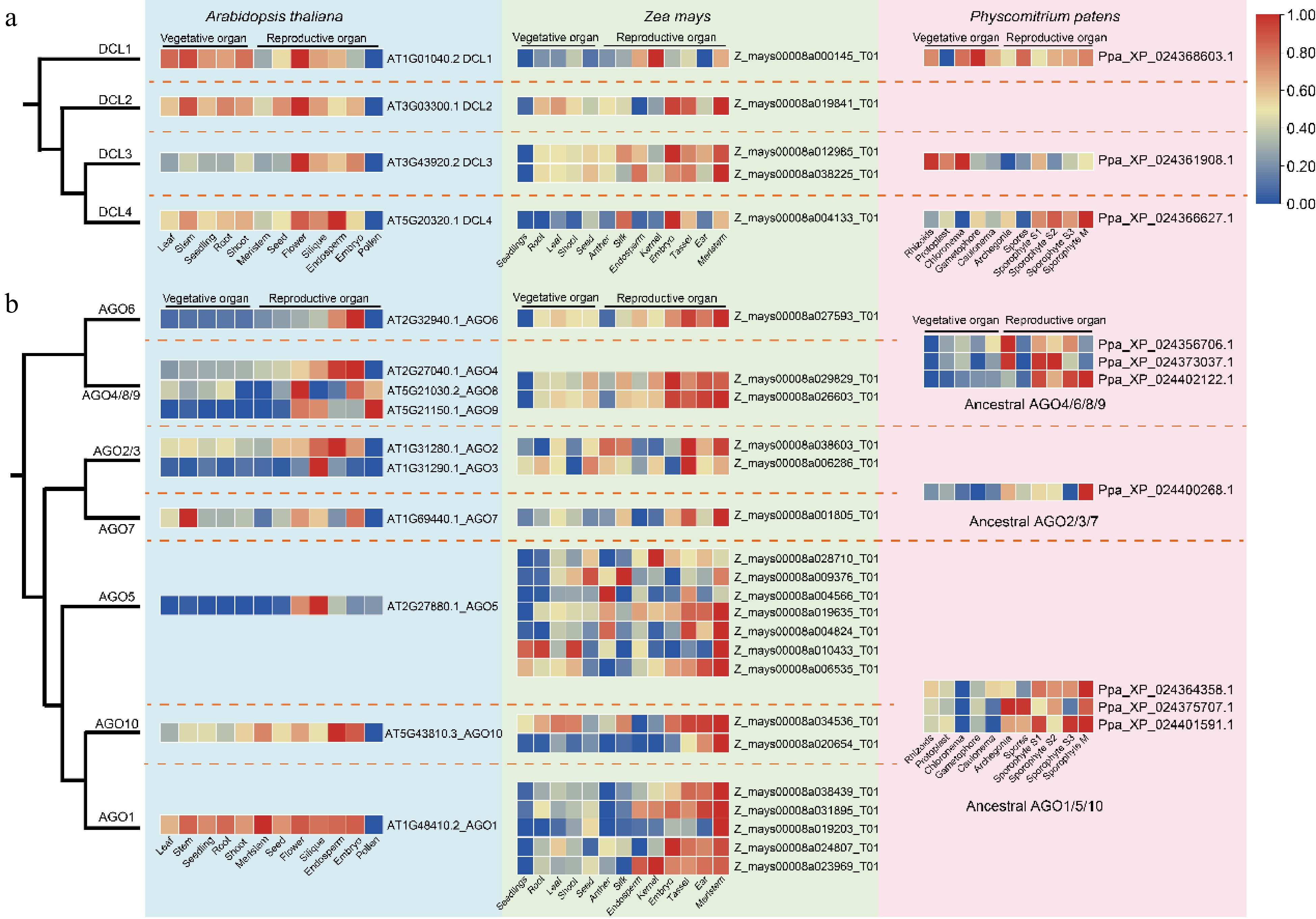
Figure 5.
Expression profiles of DCLs and AGOs in different tissues of A. thaliana, Z. may, and P. patens. (a) Comparative expression profiles of DCL gene family members. (b) Comparative expression profiles of AGO gene family members. Dashed lines demarcate distinct clades, with the heatmap displaying relative expression levels from low (blue) to high (red).
The expression of DCLs and AGOs in response to various stresses in A. thaliana and Z. may were further analyzed. The results indicate that A. thaliana DCLs respond to all stressors except irradiation, whereas Z. may DCLs primarily respond to heat, salt, drought, and nutrient deficiency (Fig. 6a). The stress responses of AGOs vary across different groups (Fig. 6b). For example, in A. thaliana, AGO6, AGO7, and AGO10 are involved in most stress pathways. In Z. mays, AGO6, AGO4/8/9, AGO2/3, and AGO10 participate in multiple stress responses. In both A. thaliana and Z. mays, AGO5 exhibits minimal responsiveness to stress, whereas AGO4 responds to similar stresses, including heat, salt, drought, cold, shade, and nutrient deficiency. This comprehensive analysis underscores the specificity and variability of DCL and AGO responses to environmental stresses, highlighting their essential adaptive functions in plant stress physiology.
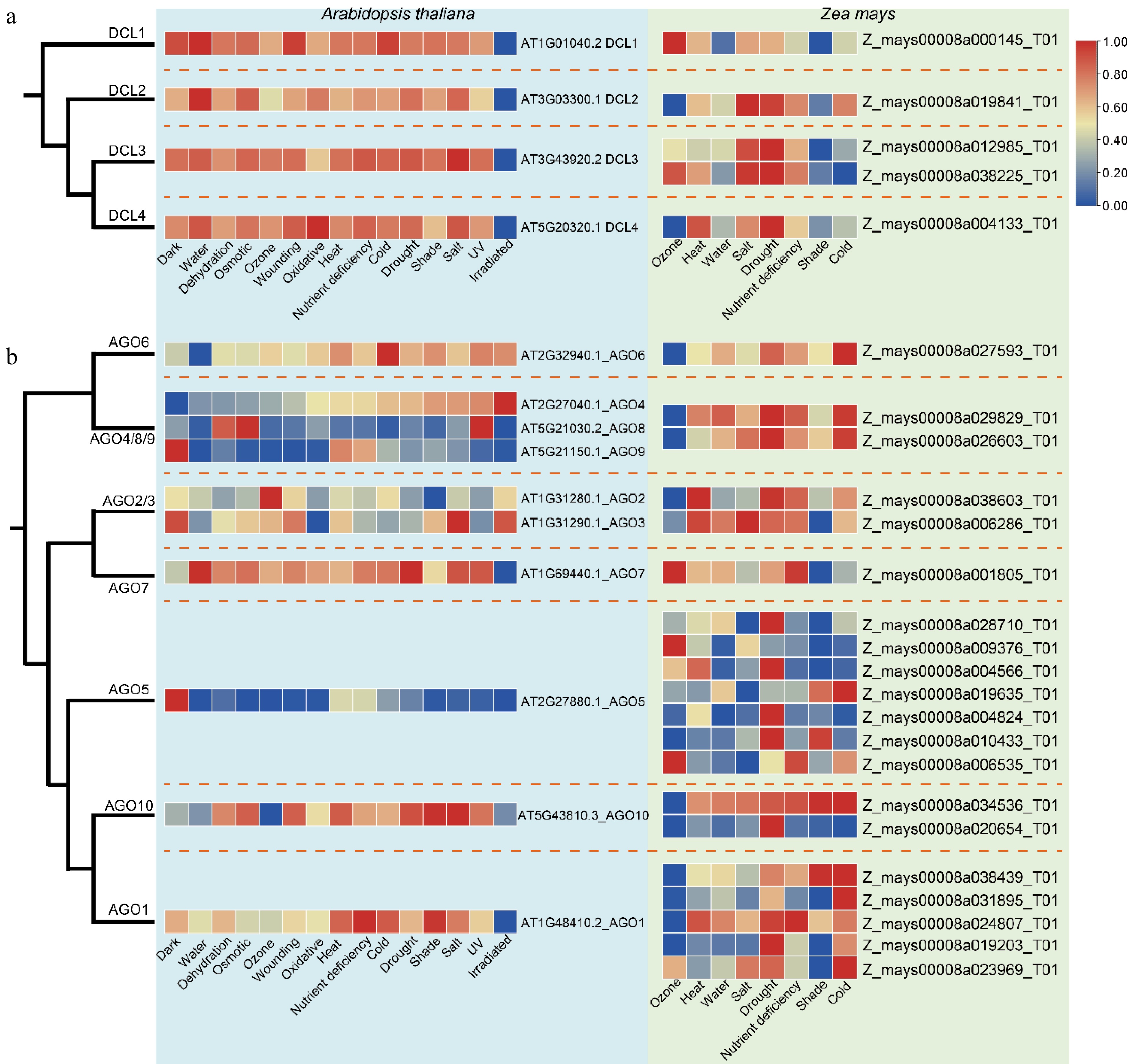
Figure 6.
Expression of the DCLs and AGOs under different stress conditions in A. thaliana and Z. mays. (a) Comparative expression profiles of DCL gene family members. (b) Comparative expression profiles of AGO gene family members. Dashed lines denote distinct clades, with the heatmap displaying relative expression levels from low (blue) to high (red).
-
As plants transitioned from aquatic to terrestrial environments, they encountered more variable habitats and increased exposure to air. This shift prompted the expansion of numerous gene families to adapt to these diverse environmental challenges[35−37]. The present findings align with previous studies, revealing that DCLs are predominantly classified into four groups, with DCL2 being exclusive to seed plants (Fig. 1a). The origin of DCLs was traced back to rhodophytes, detecting homologs in Chondrus crispus and Porphyridium purpureum, a finding supported by data from the 1KP database (Supplementary Fig. S1). Similarly, the present analysis suggests that AGOs also originated from rhodophytes, demonstrating the conservation of the RNAi pathway across plant species. Contrary to Li et al., who reported a single ancestral lineage for AGO, the present study identifies ancestral positions for AGO4/6/8/9 and AGO2/3/7 in Porphyridium purpureum[16]. Furthermore, a clear differentiation of AGOs into two distinct groups in rhodophytes, chlorophytes, and charophytes was observed (Fig. 1b). The expansion of the AGO family from charophytes to bryophytes likely represents an evolutionary adaptation crucial for terrestrial colonization[38,39]. Furthermore, these findings underscore the significant role of epigenetics in the terrestrial adaptation of plants.
DCLs exhibit a single ancestral branch in all algae, however, their expansion during the transition from aquatic to terrestrial environments coincides with that of the AGOs. This expansion continuous as lower plants evolved into higher plants, leading to the present diversification of these gene families. As key components of the RNAi mechanism, DCLs, and AGOs are crucial for the generation and function of miRNA. Specific miRNAs in algae that are conserved in seed plants, such as miR167, miR172, miR395, miR414, miR418, and miR419, are missing in mosses and ferns[40−43]. These miRNAs are essential for flower development, stress resistance, and root development in higher plants[44,45]. Their absence in mosses and ferns highlights differences in the miRNA-mediated gene silencing pathways between lower and higher plants and underscores the adaptive changes during plant evolution, reflecting species-specific survival strategies and developmental needs in diverse environments. Further analysis of the evolutionary trajectories of DCLs and AGOs in seed plants revealed that DCLs underwent two rounds of WGDs in land plants without a significant increase in their numbers (Figs 2 & 4). By contrast, AGOs experienced two WGD events and one tandem duplication (Fig. 4), and maintained specific expansions within different species (Fig. 3). These findings reveal distinct evolutionary paths of these gene families and their crucial roles in adapting RNAi mechanisms for plant survival and development across diverse ecological settings.
WGD and various forms of gene duplication are the primary mechanisms that drive the expansion of gene families. The retention of duplicated genes throughout evolution has facilitated better adaptation in plant growth and development[46−48]. Extensive research has established the functions of DCLs and AGOs. This study discovers that most DCL and AGO genes in P. patens are highly expressed in meristematic tissues, a pattern that is conserved in A. thaliana, Z. mays, and F. vesca (Fig. 5 & Supplementary Fig. S3). This expression profile is likely attributed to active DNA methylation and RNAi regulation within these tissues[10,49,50], underscoring the intricate genetic regulation essential for plant development. Additionally, DCL3 and DCL4 in P. patens show high expression in vegetative and meristematic tissues, respectively, suggesting the specialization of their functions (Figs 5 & Supplementary Fig. S3). In Arabidopsis, F. vesca, and Z. mays, the DCL2/3/4 genes are highly expressed in various tissues and response to stress (Fig. 6). The sRNAs they produce vary, indicating that diverse types of sRNAs are extensively involved in the life cycles of plants. This diversity enables plants to adapt to environmental fluctuations and supports their growth and development[51−53]. Additionally, DCL serves as the factory for manufacturing and producing miRNAs. Its tissue-specific expression, along with upregulated expression under stress conditions, is closely associated with the miRNAs it produces. For example, the miRNA156/SPL module can participate in root development and vegetative growth while also enhancing the plant's tolerance to abiotic stress. miR169 targets different members of the NF-YA gene family, which is involved in multiple developmental processes and stress responses. Moreover, miR159, miR397, and miR393 possess diverse functions in plant growth, development, and stress tolerance[44,45]. By contrast, the functions of the AGO4/6/8/9 clade have remained largely unchanged throughout evolution, with no special functions emerging from their expansion in Z. may and A. thaliana (Figs 5, 6, & Supplementary Fig. S3). Previous studies have demonstrated functional complementarity among AGO4/6/8/9 proteins[10,54,55], which is essential for maintaining critical biological processes under varying conditions. AGO2/3/7 appear to be predominantly involved in stress responses (Fig. 6), consistent with findings of previous studies[56,57]. In A. thaliana, F. vesca, and Z. mays, AGO1/5/10 exhibit significant functional divergence. Specifically, in A. thaliana, AGO1 and AGO5 exist as single-copy genes. However, AGO5 has largely lost its regulatory functions in tissue development and stress response, whereas AGO1 retains all these functions (Figs 5 & 6). In Z. mays, although there is a significant expansion of members within the AGO1/5/10 group, their functions remain largely similar to those observed in P. patens. These results suggest that subfunctionalization and neofunctionalization are two potential evolutionary outcomes of gene duplication[58], and also demonstrate the role of epigenetic regulation in directing species-specific evolutionary trajectories in plants.
-
Genomic data for 36 plant species used in this study were obtained from databases such as Phytozome (Supplementary Table S1). Gene screening and alignment of the 1KP transcriptome data were conducted using the ONEKP online platform (
https://db.cngb.org/onekp ). Protein sequences of the four DCLs and ten AGOs from A. thaliana served as query sequences for BLASTP analysis against the proteomic data of the remaining 35 plant genomes, using an e-value threshold of 1e-20. The resulting sequences were then analyzed using InterProScan to identify and annotate conserved domains using the Pfam, PANTHER, and SMART databases[59]. Only sequences that contained domains consistent with those found in A. thaliana were selected for constructing a phylogenetic tree using FastTree. Branches exhibiting abnormal lengths were manually removed to ensure the accuracy of the inferred phylogenetic relationships[60].Phylogenetic tree construction and collinearity analysis
-
First, multiple sequence alignment of all DCL and AGO protein sequences was performed using MAFFT. Gap positions were then removed from the aligned sequences using Phyutility with a cutoff parameter of 0.5. Next, ProtTest was used to predict the best-fit substitution models for constructing the DCL and AGO phylogenetic trees. The DCL and AGO trees were constructed using IQ-TREE with the JTT + F + R6 and LG + I + G + F models, respectively, with 1000 Bootstrap replicates[61]. For genes indexed in the 1KP database, phylogenetic trees were constructed using FastTree. Additionally, collinearity blocks were identified by comparing coding sequences across species using the Python version of MCScan[62].
Expression analysis of DCL and AGO genes
-
Expression data for different tissues of P. patens and F. vesca were obtained from the Physcomitrium eFP Browser and the Strawberry eFP Browser, respectively (
http://bar.utoronto.ca ). For A. thaliana and Z. mays, tissue-specific and stress-induced expression profiles were downloaded fromhttps://plantrnadb.com . Detailed data sources are provided in Supplementary Table S2. The expression of DCL and AGO genes across these species was visualized using TBtools[63]. The research was supported by the Key Research and Development Program of Jiangsu (BE2023350) and the Priority Academic Program Development of Jiangsu Higher Education Institutions Project (PAPD), and the high-performance computing platform at the Bioinformatics Center of Nanjing Agricultural University. We would like to thank A&L Scientific Editing (
www.alpublish.com ) for their linguistic assistance during the preparation of this manuscript.-
The authors confirm contribution to the paper as follows: study conception and design: Xiong AS, Cheng ZM, Su LY; data analysis, draft manuscript preparation: Su LY, Li SS, Liu H. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article.
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Li-Yao Su, Shan-Shan Li
- Supplementary Table S1 Genome data used in this study.
- Supplementary Table S2 Source of gene expression data.
- Supplementary Fig. S1 The ML tree of DCLs identified with 1KP transcriptomic data. Branches are color-coded to denote different plant groups: black for rhodophytes, yellow for chlorophytes, red for charophytes, green for ferns and bryophytes, and blue for seed plants.
- Supplementary Fig. S2 The ML tree of AGOs identified with 1KP transcriptomic data. Branches are color-coded to denote different plant groups: black for rhodophytes, yellow for chlorophytes, red for charophytes, green for ferns and bryophytes, and blue for seed plants.
- Supplementary Fig. S3 RNase III domain alignment of four groups of DCL proteins.
- Supplementary Fig. S4 MID domain alignment of three groups of AGOs.
- Supplementary Fig. S5 PAZ domain alignment of three groups of AGOs.
- Supplementary Fig. S6 Expression of DCLs and AGOs in different tissues of F. vesca. (A) Comparative expression profiles of DCL gene family members. (B) Comparative expression profiles of AGO gene family members. Dashed lines demarcate distinct clades, with the heatmap displaying relative expression levels from low (blue) to high (red).
- © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Su LY, Li SS, Liu H, Cheng ZM, Xiong AS. 2024. The origin, evolution, and functional divergence of the Dicer-like (DCL) and Argonaute (AGO) gene families in plants. Epigenetics Insights 17: e003 doi: 10.48130/epi-0024-0005 

The origin, evolution, and functional divergence of the Dicer-like (DCL) and Argonaute (AGO) gene families in plants
- Received: 12 September 2024
- Revised: 23 October 2024
- Accepted: 30 October 2024
- Published online: 28 November 2024
Abstract: Dicer-like (DCL) and Argonaute (AGO) proteins play a crucial role in plant epigenetics. However, the evolutionary origins and roles of these gene families in plant adaptation, stress response, and development remain unclear. This study explores the origin and functional evolution of DCLs and AGOs across 36 plant species spanning diverse taxonomic groups. Member identification, phylogenetic analysis, evolutionary trajectory analysis, and functional divergence analysis were conducted. The results show that the DCL and AGO originated in Rhodophytes and underwent two major expansions: during algal terrestrialization and the transition from lower to higher plants. In seed plants, DCLs diversified into four classes following two whole-genome duplication (WGD) events, whereas AGOs diversified into seven classes through two WGD events and one tandem duplication event. Expression analyses in Physcomitrium patens, Zea mays, Arabidopsis thaliana, and Fragaria vesca revealed high expression of these gene families in reproductive tissues, with notably lower expression in pollen. Additionally, the expression of these genes exhibits different responses to various environmental stresses in A. thaliana and Z. mays, highlighting their important roles in adaptation to environmental fluctuations. The present research reveals the functional diversification of DCLs and AGOs and their crucial roles in facilitating terrestrial adaptation and rapid land colonization.