









-
Papaya (Carica papaya L., Caricaceae) is a widely cultivated fruit crop in tropical and subtropical regions around the world[1]. Papaya has economic and cultural importance due to its high yield, nutritional value, and medicinal properties[2−4]. The fruit is consumed when mature as a fresh fruit or when immature as a vegetable; processed products are also produced from it. The worldwide production of papaya in 2022 was estimated to be 13,822,328 metric tons according to the Food and Agriculture Organization Corporate Statistical Database (
www.fao.org/faostat/en/#data/QCL ). In addition to its value as a food crop, the striking leaf shape and distinctive plant architecture of papaya allow this plant to be used as an ornamental plant in residential landscapes.Petiole color is an important ornamental trait in papaya. The common petiole color is green; however, there is a purple color form. The accumulation of purple pigmentation on the petiole, combined with the green lamina gives the papaya plant a unique appearance, reminiscent of a purple and green umbrella. These two distinct petiole phenotypes were first described in 1938, where purple petiole was found to be dominant over non-purple stem color[5]. Subsequent genetic analysis showed that the inheritance of stem color was fairly associated with flower colors, and loosely linked to sex type[5]. Folorunso observed that the petiole color in papaya exhibited an equal segregation ratio of 1:1 (purple : green) among the offspring that resulted from open-pollinated crosses between female and hermaphrodite papaya parents with purple petioles[6]. Interestingly, the study also revealed that the purple petiole color co-segregated with the pigment color of petals, peduncle, fruit rind, and fruit stalk[6], suggesting a genetic linkage or shared regulatory pathway controlling the pigmentation across these tissues.
In papaya, the accumulation of purple pigment is primarily attributed to a buildup of anthocyanin, which imparts the red to blue hues commonly observed in various plant tissues[7]. Anthocyanins are water-soluble natural pigments that belong to the flavonoid group, and are widely distributed across angiosperms[8]. The presence of anthocyanins is often associated with various biological functions in plants. They play a crucial role in attracting pollinators and seed dispersers, thus affecting plant reproduction rates[9]. Anthocyanins also enhance plant resilience by protecting against a range of biotic and abiotic stresses, including protection against UV light exposure[10]. Additionally, anthocyanins have been acknowledged for their antioxidant/anticarcinogenic properties and health-promoting effects in the prevention of heart disease, cardiovascular disease and cancer[11,12]. Given their importance, the pathways governing anthocyanin biosynthesis, degradation, and regulation have been extensively studied[13,14]. The biosynthesis of anthocyanins is primarily controlled by two gene groups: structural genes and regulatory genes[15]. Structural genes include those involved in the phenylpropanoid pathway, such as phenylalanine ammonia-lyase (PAL), cinnamate 4-hydroxylase (C4H), 4-coumarate:coenzyme A ligase (4CL)[16,17], which are responsible for the initial steps in the biosynthesis of flavonoids, and the genes in the flavonoid biosynthetic pathway, such as chalcone synthase (CHS), chalcone isomerase (CHI), flavanone 3-hydroxylase (F3H), 3'-hydroxylase (F3'H), flavonoid 3',5'-hydroxylase (F3'5'H), dihydroflavonol 4-reductase (DFR), leucoanthocyanidin dioxygenase (LDOX), anthocyanidin synthase (ANS), and flavonoid 3-O-glucosyltransferase (UFGT), which are active downstream of anthocyanin biosynthesis[18,19]. Regulatory genes usually influence the pattern and intensity of anthocyanin biosynthesis by controlling the expression of these structural genes. A 'MBW complex' has been widely recognized as a major regulator consisting of R2R3-MYB, basic helix–loop–helix (bHLH), and WD40 proteins[13,20−22]. These proteins can act as either activators or repressors in controlling the accumulation of anthocyanins in plants[23−25].
QTL-Seq is a highly efficient approach for rapid identification of genetic loci associated with traits of interest, offering a significant advantage over the more time-consuming and costly conventional QTL analysis methods[26]. This technique integrates bulked-segregant analysis (BSA), an elegant method to rapidly identify the specific genomic region by analyzing two bulked DNA pools consisting of F2 progeny with contrasting phenotypes using next-generation sequencing[26,27]. By comparing two bulked DNA pools representing contrasting phenotypes, the candidate genomic regions or genes are identified via the distribution of single nucleotide polymorphisms (SNPs). In addition to QTL-seq, transcriptome analysis has gained recognition as a reliable strategy for discovering genes associated with specific traits. By examining the expression patterns of genes across different tissues or stages, transcriptome analysis can provide valuable insight into the molecular mechanisms underlying phenotypic variation[28].The combination of QTL-Seq and transcriptome studies has been widely applied to identify genes associated with target traits in different plant species[29−31].
Despite two previous studies on papaya petiole color[5,6], little follow-up work has been done. However, understanding the genetic mechanism that governs petiole color in papaya is not only crucial for unraveling the fundamental biology of this trait but also has significant potential for its practical application in breeding programs. By elucidating the genetic basis of purple pigmentation in petioles, breeders could develop papaya varieties with higher aesthetic and commercial appeal. It also can provide insight into the introduction of purple pigmentation into other tissues and can contribute to the development of novel ornamental or fruit qualities, thereby increasing the economic value of papaya, optimizing plant appeal to consumers and expanding their marketability. In the present study, a joint approach combining BSA-Seq and transcriptome analysis were employed to investigate the genetic basis of petiole color in papaya. By integrating these two methods, the aim is to pinpoint specific genomic regions and the genes responsible for regulating pigmentation of petiole color in papaya. The results from this study will contribute to a deep understanding of how pigmentation is regulated in papaya, and expands the economic value of papaya through breeding new cultivars with both ornamental and fruit traits.
-
Two breeding lines, PR-2043 with green petioles and T5-2562 with purple petioles, were developed by crossing transgenic lines X17-2 with 'Tainung No. 5', and 'Puerto Rico-65' respectively[32,33], and maintained at the Tropical Research and Education Center, University of Florida, Homestead, FL, USA. PR-2043 and T5-2562 were crossed to generate an F1 population, and eight F1 of these plants were transplanted to the field. A hermaphrodite purple petiole F1 papaya plant was selfed to generate the F2 segregating population. The F2 seeds were soaked in water overnight and subsequently immersed in 2.5 mM gibberellic acid for 30 min before sowing in April 2020. These pre-treated seeds were planted in a mixture of 1:1 Promix BX mycorrhiza and perlite. Each 38-cell tray was top-dressed with Osmocote 14-14-14 fertilizer. Seedlings were maintained in the greenhouse and watered as necessary. Phenotyping of the F2 seedlings was carried out in the greenhouse three months after sowing and further confirmed in the field two months later. The petiole color was visually categorized as 'green' or 'purple', and the purple became more visible with plant growth. Chi-square analysis was conducted to assess the segregation ratio of petiole color in the F2 population.
Bulk construction, whole-genome resequencing, and SNP calling
-
The total genomic DNA was extracted from young leaves of individual F2 lines and the two parents (T5-2562 and PR-2590) following a CTAB method[34]. A total of 25 DNA samples representing each petiole color phenotype were pooled together into two DNA bulks for sequencing. Four sequencing libraries were constructed by shearing DNA into short fragments, repairing the ends, and making poly-A-tailed fragments before ligation with Illumina adapters. After size selection, quantified libraries were pooled and sequenced using a 150 bp paired-end program on Illumina HiSeq X10 platform (Novogene, Beijing, China).
Quality control of the raw sequencing reads was first determined by FastQC[35]. To ensure high-confidence variant calling, the adapters were trimmed using BBDuk[36]. The processed reads were then used to create the consensus sequences of both T5-2562 and PR-2590 by aligning to the 'SunUp' reference genome[37]. Read alignment of both F2 bulked pools were assessed by BWA software[38], and SAMtools[39]. Picard tools were used to mark duplicate and index bam files of F2 bulked pools and each parent's consensus sequences. GenomeAnalysisToolkit (GATK) was used to perform variant calling[40]. SNPs and indels were filtered by GATK VariantFiltration function with parameters QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < −12.5 || ReadPosRankSum < −8.0. Low-quality SNPs were removed from the final output, which were subsequently used for QTL analysis with the 'QTLseqr' R package[41]. The confidence intervals were determined using 10,000 simulations of the QTL-seq method as described previously[26]. The 95% (p < 0.05) confidence interval was set to consider that the genomic loci showing statistical significance[41].
Transcriptome analysis
-
The epidermal and cortex layers were collected from the papaya petiole of mature (18 months-old) PR-2043 and T5-2562 plants for the transcriptome study (< 1 mm thick). Green petioles were collected from PR-2043 and petioles in the process of turning from green to purple were collected from T5-2562. The freshly harvested tissues were flash-frozen in liquid nitrogen and then ground into fine powder for RNA extraction, two technical replications were processed for each sample. A total of 100 mg of tissue was processed with 1 mL TRIzol reagent, followed by washing with 70% ethanol and resuspension in 50 μL of DEPC-treated water. RNA-free Dnase (Qiagen, Hilden, Germany) and Rneasy PowerClean Pro Cleanup Kit (Qiagen, Hilden, Germany) were applied for further purification. The NovaSeq6000 platform was used to perform the sequencing.
Quality control and adapter removal of the raw sequence data were processed as described above. Clean reads were then mapped to the papaya reference genome using HISAT2 (--dta) before counting mapped transcripts with featureCount software following default parameters[42,43]. Genes that were differently expressed between the petioles of PR-2043 and T5-2562 were identified and quantified using DESeq2 with normalization[44]. Transcripts with a |log2(fold change)| > 2 were considered as differentially expressed genes and annotated by Blast2Go[45]. The candidate genes were identified from DEGs according to their function and false discovery rate (FDR) correction (> 0.05). The expression of candidate genes was visualized in a heatmap plotted by R Package 'Pheatmap'[46].
Quantitative PCR (qPCR)
-
To verify candidate gene expression in green and purple petioles, PR-2240 and T5-2562 were used respectively. PR-2240 is a green-petiole line genetically associated with PR-2043. The purple and green epidermal and cortex layers of petioles from mature (18 months old) T5-2562 and PR-2240 plants were collected (< 1 mm thick) freshly and frozen using liquid nitrogen, respectively. Then, high-quality RNA was extracted from collected epidermal and cortex layers by using E.Z.N.A. Plant RNA Kit (Omega Bio-tek, GA, USA). The quantity of the RNA was determined by Qubit4 (Thermo Fisher, MA, USA). The RNA samples were aliquoted to uniform concentration (744 ng/μL) and reverse transcribed into cDNA using amfiRivert Sensi cDNA Master Mix (GenDEPOT, TX, USA). The CDS nucleotide sequences of each candidate gene and Primer3 were used to develop the primer pairs for CHS, and MYB20 (Supplementary Table S1). Three biological and technical replicates were processed for each candidate gene using the housekeeping gene actin as a control. The qPCR reaction was performed in QuantStudio3 (Applied Biosystems, CA, US) in a 10 μL reaction containing 5 μL 2X PowerUp™ SYBR™ Green Master Mix (Applied Biosystems), 0.4 μM of forward and reverse primer, 3.2 μL Nuclease Free water, and 1 μL cDNA. The mixture was initially held at 50 °C for 2 min and 95 °C for 2 min, incubated at 95 °C for 15 s, followed by 40 cycles at 55 °C for 15 s, and 72 °C for 1 min. The melt curve was set at 95 °C for 15 s, 60 °C for 1 min, and 95 °C for 1 s. The 2−ΔΔCᴛ method was used to analyze the relative changes in gene expression[47].
-
To investigate the inheritance of petiole color, an F2 population was developed. By crossing PR-2043 (green petiole) × T5-2562 (purple petiole, Fig. 1a), eight F1 plants were generated and all with purple petiole. A single fruit from one of the F1 plants was used to produce the F2 seedlings used in this study. In this study, the petiole color was evaluated three months after the seed germination and then the stability of the petiole color was confirmed two months later. Of the total 280 F2 seedlings, 223 were observed to have purple petioles, and 57 had green petioles (Fig. 1b), and the purple pigmentation was observed to become more noticeable as the plant grew. The purple-to-green color segregated at a 3:1 ratio in the F2 population. The Chi-square statistic and p-value were 3.219 and 0.0728 respectively (Fig. 1c), indicating that purple petioles in papaya follow a single dominant gene inheritance model.

Figure 1.
Phenotypes of papaya petioles. (a) Petiole color of PR-2043 and T5-2562 parents. (b) Purple and green petioles of papaya F2 population. (c) Segregation of petiole color in the F2 population.
BSA-Seq analysis
-
BSA-Seq analysis was used to examine the nucleotide diversity between F2 progenies with purple petioles (F2P) and green petioles (F2G) to characterize the genomic regions responsible for papaya purple petiole color. A total of 21.4 Gb (61.02 × depth) and 29.2 Gb (83.36 × depth) sequence reads (150 bp pair end) were generated for F2P and F2G bulks using whole genome sequence (Table 1). A total of 22.4 Gb (63.87 × depth) and 29.2 Gb (83.23 × depth) raw sequence reads was generated for T5-2562 and PR-2590, respectively. Consensus genomes of each parent were constructed by using papaya 'SunUp' genome as a reference. Subsequently, SNPs calling was carried out by comparing two F2 bulks and three genomes. The short reads of F2P and F2G bulks were aligned to the two parental consensus genomes and to the 'SunUp' genome, which yielded three sets of allelic segregation with 927,513, 518,567, and 1,423,583 SNPs, respectively. SNPs with low mapping rate (< 40%) were removed from the dataset, which yielded a total of 443,996 SNPs from purple parent, 235,895 SNPs from a green parent, and 687,084 SNPs from the reference genome for QTL mapping. At a 95% confidence interval, two QTL regions (189,558−1,368,545 bp and 2,739,922−3,777,906 bp) were identified on chromosome 1 of the reference genome, two QTLs were identified on chromosome 1 of the PR-2590 consensus sequence, spanning 621,177−1,791,321 bp and 3,799,705−5,554,073 bp and one QTL was identified on chromosome 1 of the T5-2562 consensus sequence (13,715−5,961,552 bp (Fig. 2). The QTL regions consistently overlapped across the same region in chromosome 1 of all three genomes with peak QTL SNPs supported by 99% confidence levels. Genome annotation identified a total of 653 genes located in the overlapping QTL region (13,715−5,961,552 bp).
Table 1. Sequencing information of parental lines and two bulks.
Sample Raw reads Raw data Sequencing
depthEffective (%) GC (%) T5-2562 149366802 22.4 63.87 99.12 37.36 PR-2590 194642908 29.2 83.23 98.40 37.26 F2P 142694068 21.4 61.02 98.03 37.05 F2G 194944976 29.2 83.36 98.29 36.89 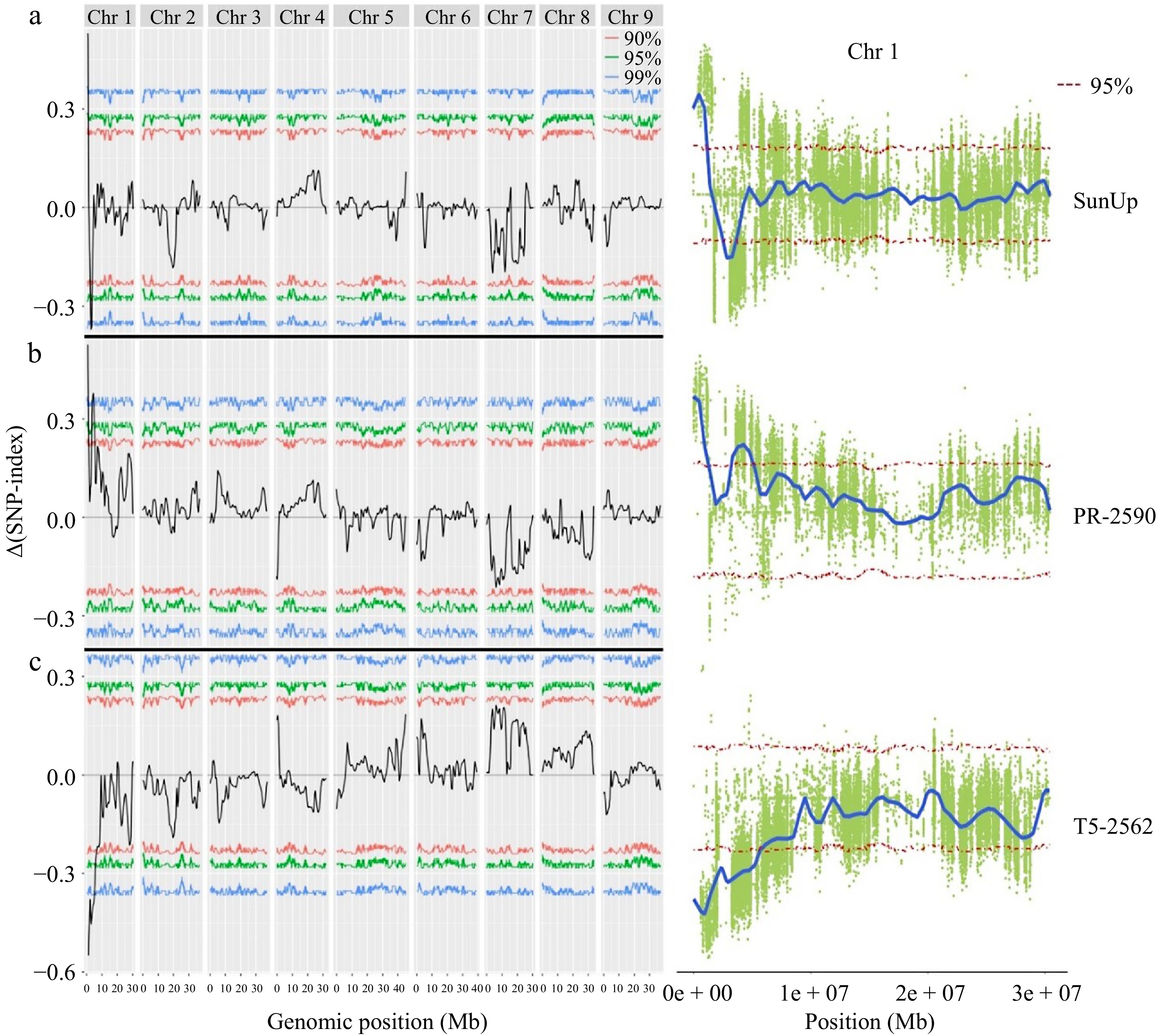
Figure 2.
QTL regions associated with papaya petiole color in three genomes, (a) SunUp, (b) PR-2590, and (c) T5-2562.
Transcriptome profiling of papaya petiole color
-
A total of 2,145 differentially expressed genes (DEGs) (|log2fold change| > 2) were identified through the transcriptome profiling of PR-2043 and from T5-2562. The GO analysis found that most DEGs were involved in various molecular functions, including small molecular binding, and organic cyclic compound binding transferase activity. Nine DEGs were involved in flavonoid biosynthetic pathways including CHI, DFR, CHS, UFGT, and flavanol synthase. Thirty-five and 17 DEGs were identified as putative MYB and bHLH transcription factors, respectively (Supplementary Table S2).
Candidate gene identification
-
The BSA-seq and transcriptome analysis identified a total of 67 genes within the QTL region on chromosome 1 that were differentially expressed between the green and purple petiole color papayas. Of them, the functional annotation identified 32 genes that acted on several biological processes, including the regulation of DNA-templated transcription, fruit ripening, methylation, and glutamine metabolism. Eleven of these play a role in molecular function, such as methyltransferase activity and nucleic acid binding. The remaining genes have a function in cellular components, including membrane, plasma membrane, and plasmodesma (Supplementary Table S3). Notably, four genes including chalcone synthase CHS, MYB315-like, MYB20, and MYB75-like, were associated with anthocyanin biosynthesis and regulation (Fig. 3a, Supplementary Fig. S1, Supplementary Table S4). CHS was highly expressed in purple petioles as compared to green petioles, suggesting CHS might play a key role in anthocyanin accumulation of papaya petioles.
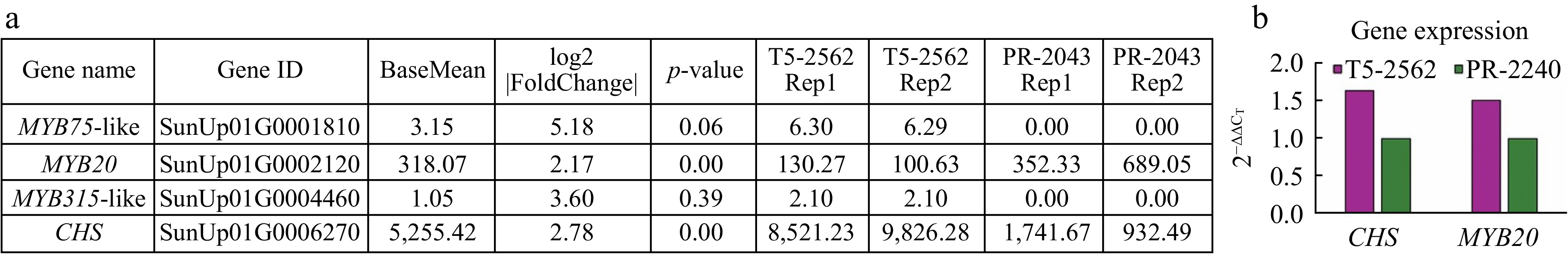
Figure 3.
Candidate genes associated with anthocyanin accumulation in papaya petiole. (a) The statistical information of candidate genes expression in different material. (b) The expression level validation of CHS and MYB20 in purple and green papaya petiole by q-PCR.
The RNA was extracted from the epidermic layer of the petiole of T5-2562 and PR-2240 to determine the expression level of CHS and MYB20 using qPCR. The qPCR results showed that the expression of CHS and MYB20 in purple petioles were both more highly expressed than that of the green petioles (Fig. 3b). The qPCR expression pattern of CHS was consistent with the RNA-seq results, whereas MYB20 showed a contradictory pattern (Fig. 3a & b; Supplementary Fig. S1). Segregation analysis, transcriptome data, and qPCR validation suggest that the MYB20 may be involved in other biological functions during petiole development, but it is not associated with petiole color in papaya.
-
Anthocyanins, water-soluble pigments generated by the phenylpropanoid pathway, contribute many pink, purple, and blue hues in plants. Anthocyanins are not only natural dyes with brilliant colors but also edible consumption that benefit heart, eye, metabolic, and cognitive health in humans[12]. The accumulation of anthocyanins contributes to pigment diversity in distinct species pigment variation. It is very common in floral tissues[48−50], and vegetative tissues[51,52]. While this within-species pigment variation is rare in displaying contrasting fruit color, like grapes[53], apples[54], and cherries[55].
Papaya is an economically and culturally important crop in the tropical areas of the world. Ornamental traits such as petiole color, leaf shape, and growth habit are value-added traits in papaya for homeowners and landscapers. In papaya, anthocyanin accumulation only appears in a few phenotypes, specifically in the epidermis of the petiole, stem, fruit stem, and leaf vein. Additionally, the purple pigmentation in the petiole was observed to become more pronounced as the papaya plant grows[6]. The present genetic study revealed that the purple phenotype is dominant over the green in papaya and follows a single dominant inheritance pattern, which is consistent with the previous hypothesis of anthocyanin accumulation in papaya[5,6]. In other species including tomatoes[56], sweet cherries[55], and blood oranges[57], a single dominant gene has also been implicated as controlling contrasting anthocyanin phenotypes. In other cases, species such as purple cabbage, however, anthocyanin accumulation is regulated by a transcription repressor[58]. Although the evidence strongly supports purple as a dominant trait in papaya petioles, the prevalence of green petiole papayas in nature remains an enigma that demands more investigation. There is evidence indicating that the inheritance of purple pigmentation in papaya stem is loosely linked to sex type[5]. Therefore, one hypothesis is that the gene governing anthocyanin accumulation in papaya was subject to human selection based on sex types during cultivation.
The anthocyanins biosynthetic pathway is downstream of the flavonoid pathway and includes structural genes such as CHS, CHI, F3H, F3'H, F3'5'H, DFR, LDOX, ANS, and UFGT[18,19]. CHI, DFR, CHS, and UFGT were found to be expressed differentially between purple and green petioles. Among these genes, CHS was the only differentially expressed gene that was also located in the QTL region identified by QTL-seq analysis. The flavonoid pathway begins when CHS mediates the synthesis of naringenin chalcone[14,15]. Several reports have indicated a positive correlation between CHS gene expression and anthocyanin content[59,60]. RNA-Seq and qPCR both verified the expression level of CHS in purple petiole is higher than that of green petiole in PR-2043 and PR-2240 compared to T5-2562, strongly suggesting that anthocyanin accumulation in papaya petiole is influenced by elevated CHS expression. MYB transcription factors have also been identified as a crucial group regulating anthocyanin biosynthesis either by acting independently on other structural genes or combining into MBW complexes with bHLH and WD40 proteins to regulate late pathway genes[61]. Contrasting anthocyanin accumulation phenotypes are often caused by mutations within the coding sequence of MYB factors, as in Chinese bayberry[62], or in the promoter region, e.g. in cauliflower[63]. In this study, a total of 35 and 17 MYB and bHLH transcripts, respectively, were detected as DEGs from the RNA-Seq analysis. Three of them lie within the QTL region identified through QTL-seq analysis. However, inconclusive expression patterns were observed in different papaya cultivars with green petioles through qPCR and RNA-Seq, suggesting further research is required to characterize the role of MYB20 in anthocyanin accumulation in papaya petioles.
Anthocyanin-rich foods, such as eggplant and blueberry are popular in the market. High-anthocyanin varieties have been developed to meet the demand for diverse and nutrient-rich produce, like blood orange, red cabbage, etc. It has been reported that the purple color pigmentation in papaya has pleiotropic effects, which is also noticed in the fruit rind, fruit stalk, and peduncle[6]. Elucidation of the genetics governing purple pigmentation in this study will not only give insight into developing the different phenotypes of papaya to explore its ornamental value but also facilitate future efforts to breed the anthocyanin-rich papaya fruits. Furthermore, the genetic mechanism behind anthocyanin accumulation in vegetative tissues can have future applications. For example, anthocyanin accumulation genes can be transformed into plants that are driven by a papaya fruit-specific promoter, to potentially develop the anthocyanin-rich fruits. CRISPR technology can also be applied to papaya for seedling selection with sex types by using anthocyanin accumulation gene as an indicator, which would greatly benefit commercial papaya growers.
This work was supported in part by the U.S. Department of Agriculture Hatch project FLA-TRC-006217.
-
The authors confirm contribution to the paper as follows: study concept and design: Chambers A, Wu X; population development and phenotyping: Brewer S, Chen S; data analysis: Chen S, Brewer S, Michael VN; manuscript preparation: Chen S, Brewer S, Michael VN, Chambers A, Wu X; manuscript revision: Chen S, Michael VN, Wu X. All authors reviewed the results and approved the final version of the manuscript.
-
The data generated is available in the Gene Expression Omnibus (GEO), NCBI, via accession number GSE269737.
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 The primers for candidate genes in papaya for qPCR.
- Supplementary Table S2 The expression level of DEGs relating to anthocyanins biosynthesis.
- Supplementary Table S3 The GO annotation of DEGs located in chromosome 1 QTL region.
- Supplementary Table S4 The NCBI blast annotation of CHS, MYB20, MYB75-like and MYB315-like.
- Supplementary Fig. S1 The differential expression heatmap of CHS, MYB20, MYB75-like and MYB315-like between purple and green petioles.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Chen S, Michael VN, Brewer S, Chambers A, Wu X. 2025. BSA-seq and transcriptome analyses reveal candidate gene associated with petiole color in papaya (Carica papaya L.). Ornamental Plant Research 5: e002 doi: 10.48130/opr-0024-0032 

BSA-seq and transcriptome analyses reveal candidate gene associated with petiole color in papaya (Carica papaya L.)
- Received: 07 June 2024
- Revised: 25 October 2024
- Accepted: 03 December 2024
- Published online: 09 January 2025
Abstract: Papaya (Carica papaya L.) is an important tropical species popular for highly nutritious fruit as well as medicinal value. In addition, non-commercial cultivation of papaya trees has resulted in dual-purpose cultivars grown for both fruit and ornamental value in residential areas. Petiole color is a key ornamental trait in papaya that varies amongst cultivars depending on anthocyanin accumulation resulting in purple or green pigmentation. Although inherited as a simple trait, genetic characterization and genomic loci responsible for the purple petiole color in papaya is unknown. In this study, F1 and F2 populations generated from two breeding lines PR-2043 (green petiole) and T5-2562 (purple petiole) were used to evaluate the inheritance patterns of petiole color as well as determine genetic loci and genes involved in petiole pigmentation in papaya through bulk segregant analysis (BSA) and transcriptome sequencing. The segregation of purple petiole color followed a single dominant gene inheritance model (3:1). BSA-seq analysis indicated key genes influencing petiole color are mainly located in chromosome 1 (0.01 to 5.96 Mb) of the papaya genome. Four major genes, including CHS, MYB20, MYB315-like, and MYB75-like within this region exhibited significant differential expression in a comparison between purple and green petiole papaya plants. A relatively high abundance of CHS transcripts was observed in purple petioles and may signify a major involvement in regulating anthocyanins accumulation in papaya petioles. The findings of this study facilitate the future efforts of breeding papaya cultivars with higher economical value in residential landscapes.
-
Key words:
- BSA-seq /
- Anthocyanins /
- Carica papaya /
- Transcriptome