










-
During the life cycles of plants, high salinity, desiccation and low temperatures are the major environmental stresses. These stresses impair the development and productivity of crops, horticultural plants and wild species, and even threaten their survival. For the perennial plants grown in frigid and temperate zones, low temperature in winter is a serious stress to overcome. Based on long term evolution, plants have developed complicated mechanisms to perceive and respond to low temperature stresses. In higher plants, cold acclimation (CA) is an important mechanism to defend against cold temperatures in winter[1]. Many physiological, biochemical, and structural changes happen during plant CA process[2, 3]. The increasing number of transcriptome analyses of gene expression at the RNA transcript level have helped us to better understand the molecular basis of CA[4, 5]. However, the transcript level of a given RNA does not always strictly correlate with the corresponding protein level in plant cells. And little has been done to elucidate the protein abundance changes during CA and de-acclimation (DA) processes[6, 7]. As a valuable approach to study the stress responses at the level of protein abundance, proteome analysis has been implemented in many studies. In Arabidopsis, putative plasma membrane proteins associated with CA were identified using a mass spectrometric approach[8]. Based on the two-dimensional difference gel electrophoresis (2-D DIGE) analysis, 26 differently expressed proteins were identified in rice, and the cellular phospholipase Dα1 protein was proven as a key candidate involved in the CA signaling pathway[9]. Balbuena et al.[10] found that the tolerant lines of sunflower showed a higher number of differentially expressed proteins in leaves, compared with freezing susceptible lines. As an important semi-permeable cellular membrane, plasma membrane plays vital roles in response to abiotic stress such as low temperature stress in plant. And plasma membrane proteins may change during CA[11]. Although many cold-stress-related proteins have been identified and analyzed, and much knowledge about CA has been added recently, the understanding of the CA mechanism is still limited, especially in woody and evergreen plants.
Tea plant [Camellia sinensis (L.) O. Kuntze] is a woody, perennial, evergreen plant and is widely planted in developing counties of the tropics and sub-tropics as an important cash crop[12]. Low temperature in winter is a key environmental factor restricting the growth of tea plants, which could lead to damage to tea plantations, decline of production, and even plant death. Therefore, understanding the tea plant CA mechanism and functional genes, and application in tea plant breeding is a crucial way to improve tea plant cold tolerance. Similar to many other plants, huge changes happen at cellular, physiological and metabolic levels during the tea plant CA process, such as the relative electrical conductivity, concentration of malondialdehyde and relative water content decrease[13, 14]. Oppositely, the palisade tissue thickness increased and the plasma membrane stability enhanced through the increasing of total proteins and unsaturated fatty acids[15]. The content of soluble sugars also increased in winter[14]. Additionally, cold induced or related genes were identified and functions were validated by different technologies in tea plant[16−20]. To further highlight the mechanisms of CA, we performed a transcriptome analysis based on RNA-seq. Many differentially expressed genes were identified and confirmed using quantitative RT-PCR analysis. These genes were grouped into signal transduction genes, cold-responsive transcription factor genes, plasma membrane stabilization related genes, osmosensing-responsive genes and detoxification genes, etc[4]. The transcriptome analysis provided a valuable chance to look into global gene expression changes at RNA level during the CA process in tea plant. Transcriptomic data are not often consistent with protein or metabolism data due to post-transcriptional modifications. So to provide new ideas towards the CA mechanism in tea plant, we examined the proteome during CA and DA processes in tea plant. Tea plant, being a broad leaved woody evergreen, may provide novel information on cold resistance mechanisms of other broad leaved evergreen plants in winter.
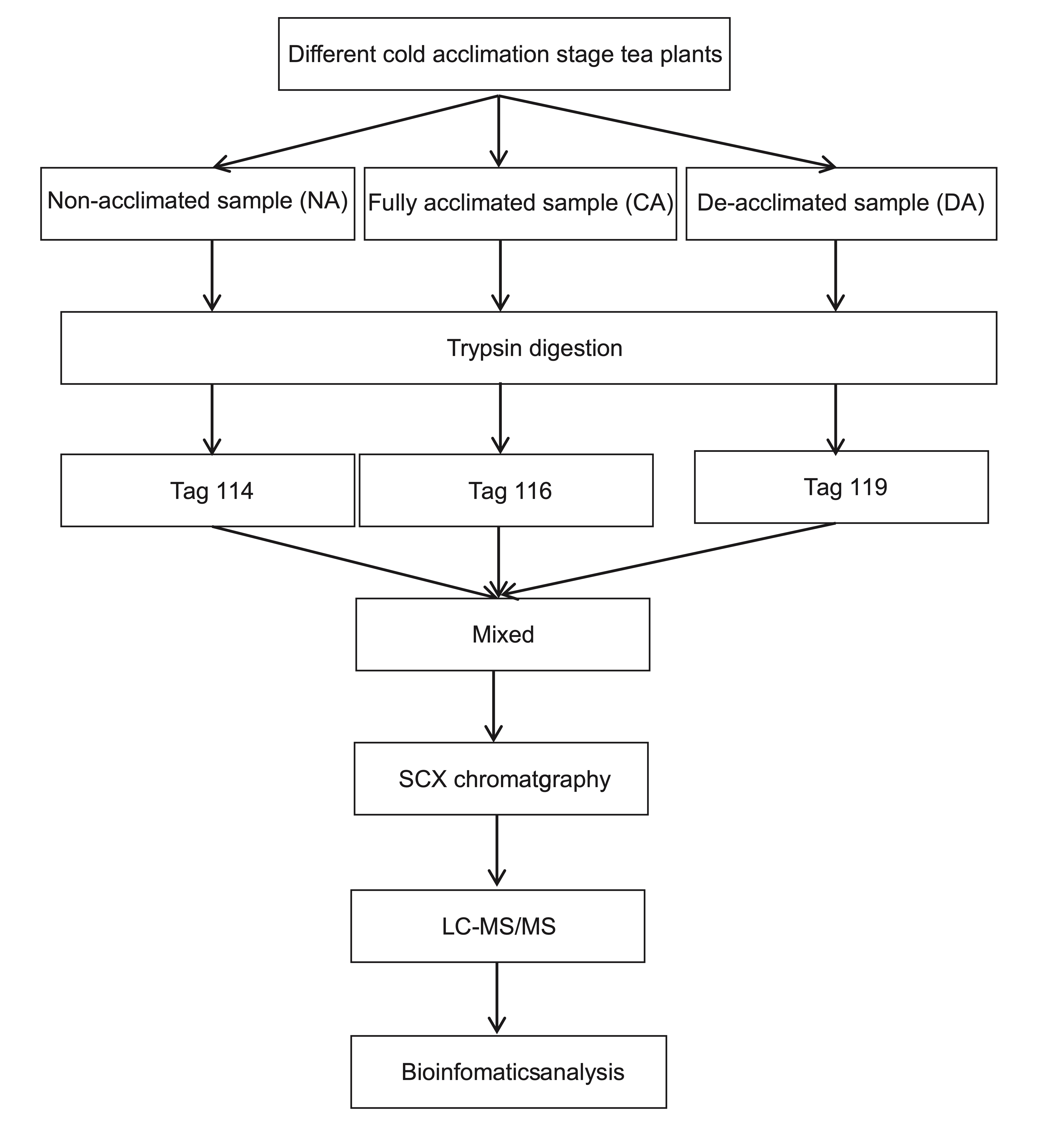
In the present study, leaf samples at the non-acclimated (NA), fully acclimated (CA) and de-acclimated (DA) stages were collected according to our previous work[4] and analyzed using isobaric tags for relative and absolute quantitation (iTRAQ) quantitative proteomic approach following the workflow shown in Supplemental Fig. S1. Finally, over 1,300 differentially expressed and functioning in varied biological processes proteins were identified.
-
The tea plants of C. sinensis (L.) O. Kuntze 'Longjing 43' grown in the field of the Tea Research Institute, Chinese Academy of Agricultural Sciences (TRI, CAAS) (N 30°10', E 120°5'), were used in this study. Intact mature leaves at NA, CA and DA stages were collected for further study according to Wang et al.[4]. Each biological replicate contained ten intact leaves collected from ten individual plant and three biological replicates of each stage were collected. Those collected samples were frozen in liquid N2, and stored at −80 °C for protein extraction and iTRAQ assay and qRT-PCR analysis.
Protein extraction and gel eletrophoresis
-
Protein extraction was performed according to the method of Yang et al.[21] with minor revision. Frozen leaves were ground to a fine power and weighted 1.0 g, and then 5 ml lysis buffer (7 M Urea, 2 M Thiourea, 4% CHAPS, 40 mM Tris-HCl, pH 8.5) was added. After that, 1 mM and 2 mM PMSF and EDTA were added respectively. Five minutes later, DTT was added with a final concentration of 10 mM. Then the above suspension was sonicated (200 Watts) for 15 min and centrifuged (30,000× g) at 4 °C for 15 min. Then those new supernatant was transferred into another tube and five-fold 10% chilled TCA acetone was added and incubated overnight at −20 °C. The precipitate was washed three times with chilled acetone for at least 30 min, and harvested by a centrifugation at 4 °C, 30,000× g for 15 min after each washing. The pellet was air dried before being dissolved in 500 μl 0.5 M TEAB, followed by sonication at 200 Watts for 15 min. Sonicated solution was centrifuged at 4 °C, 30,000× g for 15 min. Finally, supernatant was transferred to a new tube and quantified using BSA as standard protein with GE Healthcare's 2-D Quant Kit (Code No. 80-6483-56) following the instructions. After the quantification, SDS-PAGE and standard colloidal staining were performed to measure the quality of the protein sample.
In gel trypsin digestion, iTRAQ labeling and LC-MS/MS analyses
-
One hundred micrograms of protein were digested using Trypsin Gold (Promega, Madison, WI, USA) with a ration of protein : trypsin = 30 : 1, and incubated at 37 °C for 16 h. Then, the digested proteins were dried using vacuum centrifugation and reconstituted in 0.5 M TEAB (Triethylammonium bicarbonate buffer). The reconstituted NA, CA and DA samples were labeled with different isobaric tags according to the munufacturer's protocol for 8-plex iTRAQ (Applied Biosystems).
The labeled peptides were separated using Strong Cation Exchange Choematography (SCX), and analyzed using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS)[10]. Shimadzu LC-20AB HPLC Pump system was used for SCX chromatography as follows: the digested peptides were dissolved with 4 mL buffer A (25 mM NaH2PO4 in 25% ACN, pH 2.7) and then were loaded onto a Ultremex SCX column (4.6 mm × 250 mm) containing 5 μm particles (Phenomenex). Parameters of peptide separating procedures were setted as follows: first buffer A for 10 min, then 5%−35% buffer B (25 mM NaH2PO4, 1 M KCl in 25% ACN, PH 2.7) for 40 min, and thirdly 35%−80% buffer B for 1 min, with a flow rate 1 mL/min. The absorbance at 214 nm was monitored and separated samples were collected every 1 min. Finally, each sample were separated into 20 fractions, then each fraction was desalted using Strata XC18 column (Phenomenex) and dried by vacuum.
The desalted and dried fractions were re-suspended using buffer A (2% ACN, 0.1% FA) and centrifuged (20,000× g) for 10 min. Then, 8 μl of 0.5 μg·μl−1 supernatant was injected into a Shimadzu LC-20AD nano HPLC system with an analytical C18 column (inner diameter 75 μm). The flow rate was as follows: 8 μL·min−1 for 4 min, 300 μL·min−1 for 40 min with 2% to 35% B (95% ACN, 0.1% FA), followed by 5 min linear gradient to 80%, and mainted for 4 min with 80% B, finally returned to 5% within 1 min.
After that, the samples were nanoelectrospray ionized (1.6 kV) and put into an Q EXACTIVE (Thermo Fisher Scientific, San Jose, CA, USA) coupled online with HPLC system. And the high-energy collision dissociation (HCD) operating mode with a normalized collision energy setting of 27.0 was applied for peptides selected for MS/MS. An Orbitrap with a resolution of 70,000 automatic gain control (AGC) was applied for detection and spectra optimization. For AGC target, the parameters of MS and MS 2 were setted as 3e6 and 1e5 respectively. While for MS and MS 2 scans, the m/z parameters were setted as 350 to 2,000 Da and 100−1,800 Da respectively. The 15 most abundant precursor ions which have a threshold ion count above 20,000 and 15 s dynamic exclusion duration time were identified based on a data-dependent procedure.
Protein identification, bioinfomatics and differential abundance protein analysis
-
Proteome Discoverer 1.2 (PD 1.2, Thermo Fisher Scientific) was used for the conversion of raw data files acquired from the Orbitrap and the converted MGF file was used for protein identification using Mascot 2.3.02 (www.matrixscience.com) software (Matrix Science, London, UK). Tea plant unigene (from NCBI) translation database, theaceae_txid27065 database in NCBInr, and sequences from the tea plant transcriptome studies implemented by Wang et al.[4] were chosen as databases. The mass tolerance was set as '± 0.05 Da' and '± 0.1 Da' for intact peptide and fragmented ion identification respectively and one missed cleavage was allowed in trypsin digests. The charge states of peptides were set to +2 and +3. Deamidated (NQ), Gln->pyro-Glu (N-term Q) and Oxidation (M) were deemed as potential variable modifications. Moreover, Carbamidomethyl (C), iTRAQ8plex (K) and iTRAQ8plex (N-term) were defined as fixed modifications. A random sequence of database and the real database were used for raw spectra test in the decoy checkbox. An automatic decoy database search was conducted based on the decoy checkbox in Mascot. To improve the accuracy of peptide identification, only those peptides with significance scores (≥ 20) at the 95% confidence interval and detected as greater than 'identity' by Mascot probability analysis could be counted as identified. At least one unique peptide should be identified for each confident protein[22].
Protein amount was estimated by spectral counts according to Balbuena et al.[10] A pairwise comparison was performed for NA, CA and DA and 1.5 fold cutoff wit p-value < 0.05 was defined as up-accumulated or down-accumulated proteins.
Functional classification, protein abundance profiling and association analysis
-
All identified proteins were classified using standard Gene Ontology (GO) online tool (
www.geneontology.org ) analysis. The enriched GO terms among the comparisons were identified using the statistical method described by Zheng & Wang[23]. The KEGG pathways analysis was carried out by sequence alignment against the Kyoto Encyclopedia of Genes and Genomes database[24] using BLASTP algorithm (E-value threshold 10−5). Differentially accumulated protein species among the different samples were grouped by Cluster 3.0 software and the output files were read by javaTreeview.To detect the correlation between protein level and corresponding transcript level at different CA stages, all the identified protein species were matched to the corresponding transcripts in the transcriptome database[4], and then correlation analyses were implemented according to Zheng et al.[25].
Quantitative real-time PCR (qRT-PCR) analysis
-
RNAprep pure Plant Kit (Tiangen, Beijing, China) was used for total RNA extraction from the samples that were used for protein extraction according to the manufacturer's instructions. RNA concentration and integrity estimation, cDNA synthesis and real-time PCR were performed according to previous descriptions[4], and the polypyrimidine tract-binding protein gene of the tea plant (CsPTB) was used as an internal reference[26]. The gene-specific primers for qRT-PCR were designed according to the corresponding coding sequences in the genome database[4], and all the primer sequences were provided in Supplemental Table S1.
-
During the tea plant CA procedure, its relative electrical conductivity following subsequent freezing decreased, then its cold tolerance could be improved. When tea plant was de-acclimated, its relative electrical conductivity increased and its cold tolerance became weaker[4]. We collected tea plant leaves from three different stages, NA, CA and DA for iTRAQ assaies.
In total, 2,573 unique peptides out of 2,751 peptides were harvested. Based on Mascot searching in tea plant unigene translation database, NCBInr, theaceae_txid27065 database, and 51,940 sequences database 1,331 proteins were identified. Peptide length was generally 7 to 16 amino acids, and protein mass distribution was concentrated mainly at 10 to 40 kDa. Approximately 25% proteins had 5%−10% coverage by peptides, and 712 of 1,331 identified proteins were only represented by a single peptide (Supplemental Table S2). Above analyses suggested that high quality protein abundance libraries with low redundancy were constructed successfully.
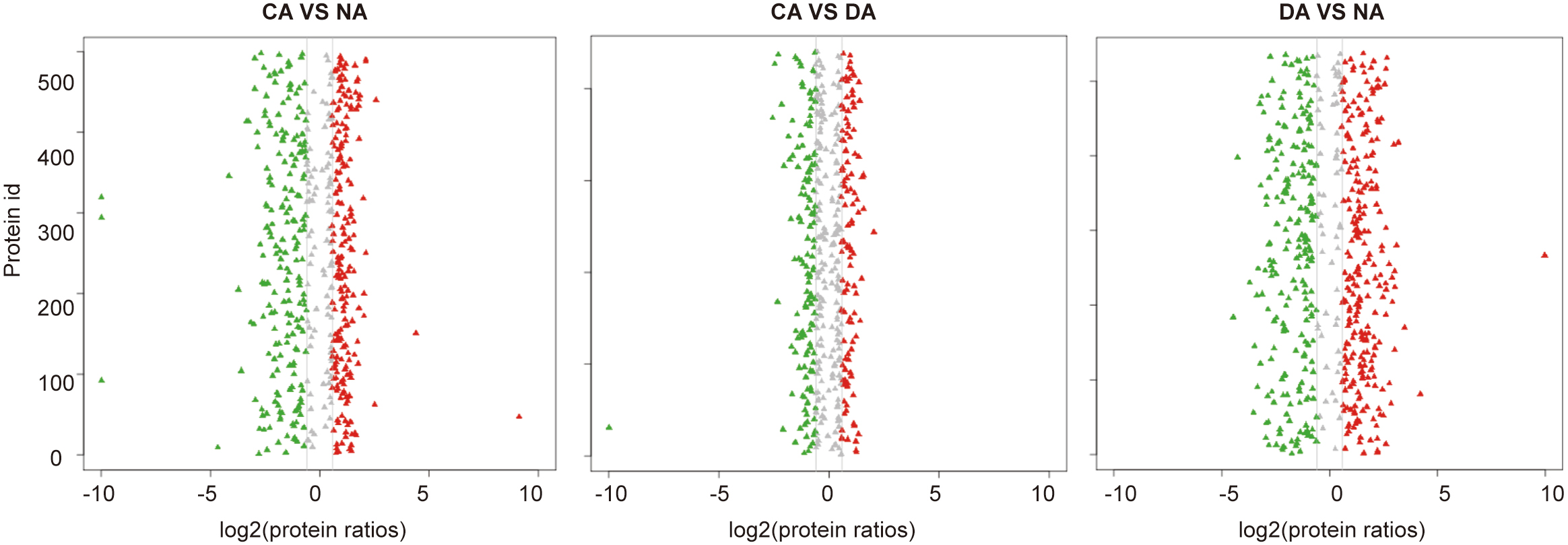
Protein quantification revealed 407 differential accumulated proteins between CA compared with NA, and among those proteins 202 were up-accumulated, while 205 were down-accumulated. Compared with CA, 115 up-accumulated and 136 down-accumulated proteins were detected in DA. In addition, compared with NA, 477 differentially accumulated proteins, including 253 up-accumulated proteins and 224 down-accumulated proteins, were identified in DA (Fig. 1). The distribution analysis of differential abundance proteins indicated that, when comparing CA with NA samples, generally down-accumulated proteins had greater abundance differences. When comparing DA with NA samples, both up-accumulated proteins and down-accumulated proteins had large abundance difference (Supplemental Fig. S2).
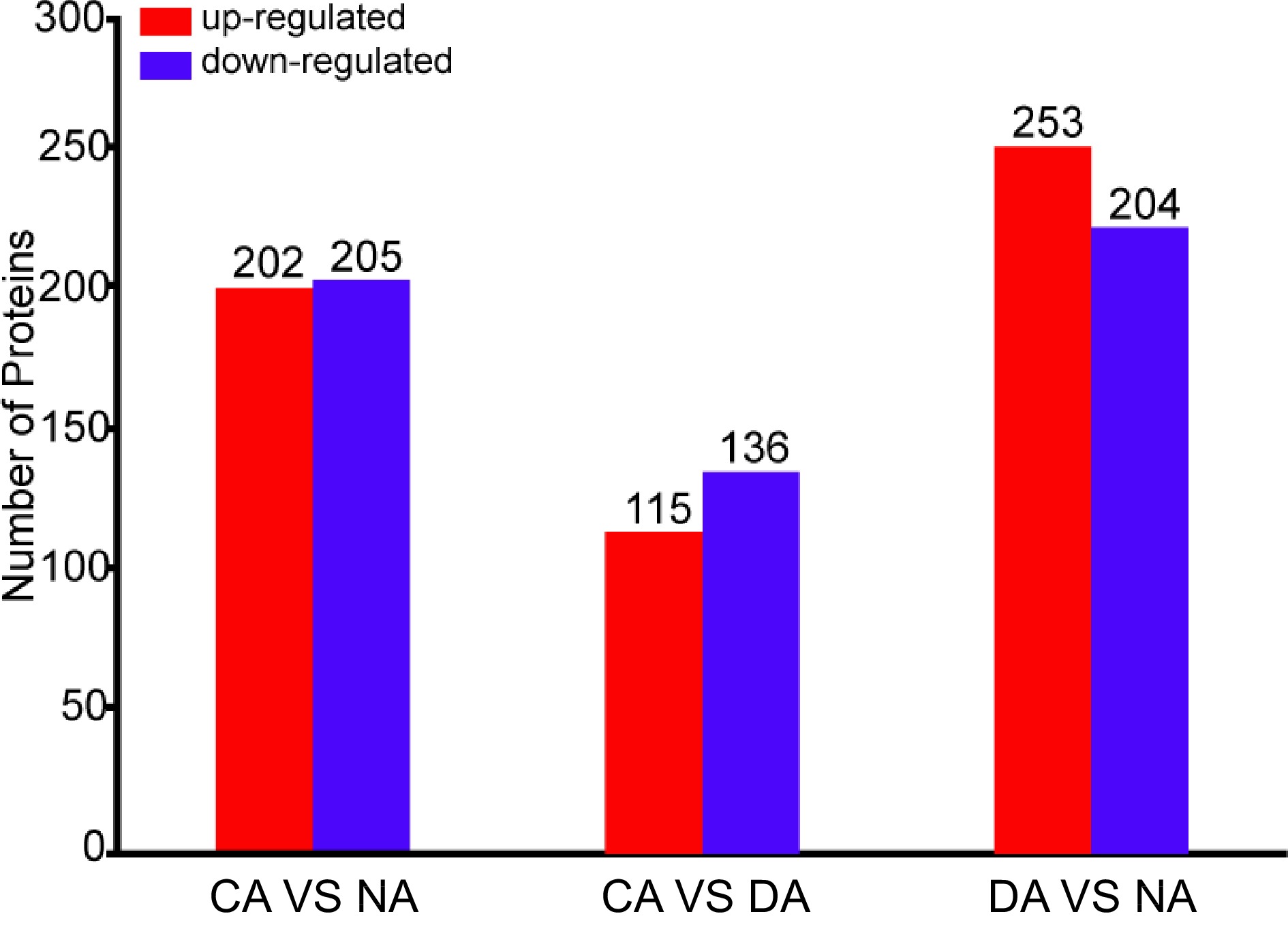
Figure 1.
Number of differentially accumulated proteins among different samples.
GO enrichment analysis of differential abundance protein species
-
Based on the GO enrichment analysis, those differently enriched GO terms (P-value < 0.05) among the comparisons between NA, CA and DA were listed in Table 1. Most of the significant differently enriched GO terms were grouped in biological process in the comparisons. Moreover, in cellular component category, more enriched GO terms were detected in DA vs CA and DA vs NA than in CA vs NA. Similar numbers of enriched GO term were grouped in molecular function category among the three different comparisons. Briefly, in the comparison of CA vs NA, extracellular region, plastid stroma, ammonia ligase activity, acid-ammonia (or amide) ligase activity, ribosome biogenesis and glutamine metabolic process were the most highly enriched; in DA vs CA, cell wall, plastid part, adenylyltransferase activity, racemase and epimerase activity, electron transport chain, and oxidation reduction were primarily enriched; in DA vs NA, extracellular region, cell wall, binding, protein kinase activity, electron transport chain and oxidation reduction were mainly enriched. Furthermore, comparing all the enriched GO terms in CA vs NA with DA vs CA, only one common enriched GO term, plastid stroma, was found. Six terms were found when comparing CA vs NA and DA vs NA, namely acid-ammonia (or amide) ligase activity, extracellular region, ammonia ligase activity, ribosome biogenesis, binding, glutamine metabolic process. Fourteen terms were found in the comparison of DA vs CA and DA vs NA, including cell wall, oxidation reduction, nucleoside phosphate metabolic process, electron transport chain, photosynthetic electron transport chain, nucleobase, nucleoside and nucleotide metabolic process, nucleotide metabolic process, photosynthesis, light reaction, purine nucleotide metabolic process, oxidoreduction coenzyme metabolic process, pyridine nucleotide metabolic process, nicotinamide nucleotide metabolic process, photosynthesis, and generation of precursor metabolites and energy.
Table 1. Gene Ontology (GO) enrichment analysis of differentially accumulated protein species among the comparisons between sample NA, CA and DA ( p -value < 0.05).
GO term NA vs CA CA vs DA DA vs NA Cellular component Extracellular region Cell wall Extracellular region Plastid stroma Plastid part Cell wall Cytoplasmic vesicle Plastid stroma Plastid envelope Vesicle Thylakoid light-harvesting complex External encapsulating structure Chloroplast thylakoid membrane Organelle envelope Light-harvesting complex Envelope Plastid thylakoid membrane Microbody Chromosome Membrane part Molecular function Ammonia ligase activity Adenylyltransferase activity Binding Acid-ammonia (or amide) ligase activity Racemase and epimerase activity Protein kinase activity Oxidoreductase activity, acting on the
CH-NH2 group of donors, disulfide as
acceptorO-acyltransferase activity Ammonia ligase activity Ligase activity, forming carbon-nitrogen
bondsRacemase and epimerase activity, acting
on carbohydrates and derivativesAcid-ammonia (or amide) ligase activity Binding Biological process Ribosome biogenesis Electron transport chain Electron transport chain Glutamine metabolic process Oxidation reduction Oxidation reduction Cellular carbohydrate metabolic process Nucleoside phosphate metabolic process Photosynthetic electron transport chain Reproductive developmental process Water-soluble vitamin metabolic process Nucleotide metabolic process Reproductive process Nucleobase, nucleoside and nucleotide metabolic process Generation of precursor metabolites and energy Glycine metabolic process Photosynthetic electron transport chain Ribosome biogenesis Cellular amino acid metabolic process Nucleotide metabolic process Nucleoside phosphate metabolic process Cellular amine metabolic process Seed germination Glutamine metabolic process Sulfur amino acid metabolic process Mucilage metabolic process Nucleobase, nucleoside and nucleotide metabolic process Reproductive structure development Glucan metabolic process Negative regulation of molecular function Carbohydrate metabolic process Vitamin metabolic process Protein complex assembly Reproduction NADP metabolic process Oxidoreduction coenzyme metabolic process Amine metabolic process NADPH regeneration Tissue development Respiratory electron transport chain Nicotinamide metabolic process Pyridine nucleotide metabolic process Flower development Alkaloid metabolic process Cellular macromolecular complex assembly Organic acid catabolic process Nucleobase, nucleoside, nucleotide and nucleic acid metabolic process Cellular protein complex assembly Carboxylic acid catabolic process Cellular glucan metabolic process Nicotinamide nucleotide metabolic process Response to chemical stimulus Photosynthesis, light reaction Protein complex biogenesis Response to inorganic substance Purine nucleotide metabolic process Cellular component biogenesis Energy derivation by oxidation of organic compounds Oxidoreduction coenzyme metabolic
processCellular component organization Organic acid metabolic process Pyridine nucleotide metabolic process Sulfur metabolic process Carboxylic acid metabolic process Nicotinamide nucleotide metabolic process Primary metabolic process Cellular ketone metabolic process Cellular polysaccharide metabolic process Photosynthesis Oxoacid metabolic process Glycogen metabolic process Purine nucleotide metabolic process Sulfur amino acid biosynthetic process Energy reserve metabolic process Fatty acid metabolic process Serine family amino acid metabolic process Photosynthesis Photosynthesis, light reaction Generation of precursor metabolites and energy Plastid membrane organization Stomatal movement Membrane organization KEGG pathway mapping
-
Pathway analysis is an important approach to expose the crucial biochemical metabolism and signal transduction pathways including given proteins[27]. The identified protein species in this study were annotated based on KEGG database. Generally, more differential abundance protein species were annotated and assigned to larger number of pathways in CA vs NA and DA vs CA, compared with DA vs CA. Moreover, the differential abundance protein species in three different comparisons were mainly mapped onto carbon fixation in photosynthetic organisms, metabolic pathway, ribosome, starch and sucrose metabolism, biosynthesis of secondary metabolites and microbial metabolism in diverse environments, protein processing in endoplasmic reticulum, photosynthesis, plant-pathogen interaction and oxidative phosphorylation. Interestingly, the pathways of glycolysis/gluconeogenesis, starch and sucrose metabolism and pyruvate metabolism related to glycometabolism were the largest proportion of differential abundance protein species in CA vs NA and DA vs NA. In addition, lysosome, glutathione metabolism, peroxisome and ascorbate and aldarate metabolism and phagosome pathways had dramatic difference in the number of differential abundance protein species in the three comparisons. As listed in Table 2 and Supplemental Tables S3 & S4, many pathways only had detectable changes in one or two specific samples, for example, proteasome, fatty acid metabolism, streptomycin biosynthesis, biosynthesis of unsaturated fatty acids and so on.
Table 2. Pathway analysis of total proteins and enriched proteins in the comparisons among different samples.
Number Pathway Count Pathway ID Total
(1,015)CA vs NA
(319)CA vs DA
(196)DA vs NA
(362)1 Metabolic pathways 458 149 91 168 ko01100 2 Biosynthesis of secondary metabolites 247 75 33 74 ko01110 3 Microbial metabolism in diverse environments 190 70 32 73 ko01120 4 Ribosome 83 24 17 27 ko03010 5 Carbon fixation in photosynthetic organisms 71 33 13 31 ko00710 6 Glycolysis / Gluconeogenesis 65 22 5 25 ko00010 7 Methane metabolism 53 17 8 21 ko00680 8 Starch and sucrose metabolism 52 19 10 24 ko00500 9 Protein processing in endoplasmic reticulum 49 17 11 15 ko04141 10 Lysosome 49 11 10 16 ko04142 11 Pyruvate metabolism 48 18 7 20 ko00620 12 Amino sugar and nucleotide sugar metabolism 43 11 8 14 ko00520 13 Photosynthesis 43 13 13 17 ko00195 14 Glutathione metabolism 36 7 7 14 ko00480 15 Phenylpropanoid biosynthesis 34 11 7 13 ko00940 16 Alanine, aspartate and glutamate metabolism 31 12 7 11 ko00250 17 Citrate cycle (TCA cycle) 31 10 3 10 ko00020 18 Plant-pathogen interaction 30 12 7 15 ko04626 19 Pentose phosphate pathway 30 13 6 12 ko00030 20 Glycine, serine and threonine metabolism 29 13 6 13 ko00260 21 Antigen processing and presentation 29 10 5 8 ko04612 22 Oxidative phosphorylation 28 11 8 12 ko00190 23 Glyoxylate and dicarboxylate metabolism 26 12 5 11 ko00630 24 Purine metabolism 25 7 4 7 ko00230 25 Huntington's disease 25 9 4 5 ko05016 26 Phenylalanine metabolism 25 10 8 10 ko00360 27 Fructose and mannose metabolism 25 8 8 7 ko00051 28 Peroxisome 23 7 4 9 ko04146 29 Galactose metabolism 22 4 5 8 ko00052 30 Arginine and proline metabolism 22 7 3 5 ko00330 31 Phagosome 21 3 5 11 ko04145 32 Nitrogen metabolism 21 13 10 13 ko00910 33 Cysteine and methionine metabolism 19 7 1 6 ko00270 34 Spliceosome 19 6 3 6 ko03040 35 Other glycan degradation 19 6 3 7 ko00511 36 Alzheimer's disease 18 8 3 8 ko05010 37 Valine, leucine and isoleucine degradation 17 4 1 7 ko00280 38 Parkinson's disease 16 5 3 3 ko05012 39 Butanoate metabolism 16 5 2 5 ko00650 40 Ascorbate and aldarate metabolism 16 7 3 4 ko00053 41 RNA degradation 16 5 2 6 ko03018 42 Pentose and glucuronate interconversions 15 7 4 7 ko00040 43 Glycerolipid metabolism 15 6 3 7 ko00561 44 Endocytosis 13 5 4 3 ko04144 45 Toxoplasmosis 13 5 3 3 ko05145 46 Propanoate metabolism 13 5 2 3 ko00640 47 Phenylalanine, tyrosine and tryptophan biosynthesis 13 3 3 2 ko00400 48 Aminoacyl-tRNA biosynthesis 13 4 1 4 ko00970 49 Cyanoamino acid metabolism 13 8 2 6 ko00460 50 Renin-angiotensin system 13 3 5 6 ko04614 51 Aminobenzoate degradation 12 4 4 6 ko00627 52 Inositol phosphate metabolism 12 4 2 4 ko00562 53 RNA transport 12 7 2 7 ko03013 54 Valine, leucine and isoleucine biosynthesis 12 3 1 2 ko00290 55 Selenoamino acid metabolism 11 3 3 4 ko00450 56 Tyrosine metabolism 11 6 3 4 ko00350 57 Tropane, piperidine and pyridine alkaloid biosynthesis 11 3 5 1 ko00960 58 Proteasome 11 2 2 ko03050 59 Reductive carboxylate cycle (CO2 fixation) 11 1 3 ko00720 60 Tryptophan metabolism 10 3 1 5 ko00380 61 Two-component system 10 6 5 8 ko02020 62 MAPK signaling pathway 10 4 3 2 ko04010 63 Fatty acid metabolism 10 4 6 ko00071 64 Porphyrin and chlorophyll metabolism 9 2 2 ko00860 65 beta-Alanine metabolism 9 3 1 2 ko00410 66 Glycosphingolipid biosynthesis - globo series 9 5 1 5 ko00603 67 alpha-Linolenic acid metabolism 9 1 1 4 ko00592 68 Terpenoid backbone biosynthesis 9 1 1 2 ko00900 69 Prion diseases 9 7 2 3 ko05020 70 Type I diabetes mellitus 9 3 2 ko04940 71 Pyrimidine metabolism 9 1 1 ko00240 72 One carbon pool by folate 9 5 3 4 ko00670 73 Flavonoid biosynthesis 8 4 1 2 ko00941 74 Chagas disease 8 3 4 ko05142 75 Lysine biosynthesis 8 1 1 2 ko00300 76 Insulin signaling pathway 8 2 1 2 ko04910 77 Chloroalkane and chloroalkene degradation 8 2 1 2 ko00625 78 Carotenoid biosynthesis 8 2 1 3 ko00906 79 Pathogenic Escherichia coli infection 8 1 4 6 ko05130 80 Proximal tubule bicarbonate reclamation 8 1 2 2 ko04964 81 Glycosphingolipid biosynthesis - ganglio series 7 2 2 3 ko00604 82 PPAR signaling pathway 7 2 3 ko03320 83 Limonene and pinene degradation 7 3 3 ko00903 84 Glycosaminoglycan degradation 7 2 2 3 ko00531 85 Metabolism of xenobiotics by cytochrome P450 7 3 4 3 ko00980 86 Sphingolipid metabolism 7 3 3 4 ko00600 87 Glycerophospholipid metabolism 7 3 1 3 ko00564 88 Vibrio cholerae infection 7 2 4 ko05110 89 Drug metabolism - cytochrome P450 7 3 4 3 ko00982 90 Lysine degradation 7 3 3 ko00310 91 Amyotrophic lateral sclerosis (ALS) 7 4 2 4 ko05014 92 Calcium signaling pathway 6 1 1 ko04020 93 NOD-like receptor signaling pathway 6 3 2 3 ko04621 94 Protein digestion and absorption 6 4 2 ko04974 95 Isoquinoline alkaloid biosynthesis 6 3 2 1 ko00950 96 Pathways in cancer 6 3 2 3 ko05200 97 Prostate cancer 6 3 2 3 ko05215 98 Neurotrophin signaling pathway 6 2 4 ko04722 99 Folate biosynthesis 6 1 3 3 ko00790 100 Ubiquinone and other terpenoid-quinone biosynthesis 6 2 3 ko00130 101 Protein export 5 2 2 ko03060 102 Photosynthesis - antenna proteins 5 2 2 2 ko00196 103 Histidine metabolism 5 2 2 ko00340 104 Riboflavin metabolism 5 4 2 2 ko00740 105 Sulfur metabolism 5 1 2 1 ko00920 106 Streptomycin biosynthesis 5 2 ko00521 107 Benzoate degradation 5 2 3 ko00362 108 Bisphenol degradation 5 1 1 1 ko00363 109 MAPK signaling pathway - yeast 5 1 2 2 ko04011 110 Arachidonic acid metabolism 5 1 1 1 ko00590 111 Progesterone-mediated oocyte maturation 5 3 1 2 ko04914 112 Type II diabetes mellitus 5 1 1 ko04930 113 Biosynthesis of ansamycins 5 4 2 4 ko01051 114 Chlorocyclohexane and chlorobenzene degradation 5 2 2 ko00361 115 Novobiocin biosynthesis 5 2 1 1 ko00401 116 Fatty acid biosynthesis 4 1 2 ko00061 117 Polycyclic aromatic hydrocarbon degradation 4 3 1 1 ko00624 118 Linoleic acid metabolism 4 1 ko00591 119 Systemic lupus erythematosus 4 1 2 ko05322 120 Bacterial invasion of epithelial cells 4 2 1 ko05100 121 Regulation of actin cytoskeleton 4 2 2 3 ko04810 122 Biosynthesis of unsaturated fatty acids 4 2 ko01040 123 Geraniol degradation 4 1 2 ko00281 124 Focal adhesion 4 1 2 1 ko04510 125 Collecting duct acid secretion 4 3 ko04966 126 Pantothenate and CoA biosynthesis 4 1 ko00770 127 Epithelial cell signaling in Helicobacter pylori infection 4 3 ko05120 128 Oocyte meiosis 4 1 3 ko04114 129 Cell cycle 4 1 2 ko04110 130 Fluorobenzoate degradation 3 1 ko00364 131 Gap junction 3 2 3 ko04540 132 Toll-like receptor signaling pathway 3 1 1 ko04620 133 N-Glycan biosynthesis 3 1 ko00510 134 Taurine and hypotaurine metabolism 3 1 1 1 ko00430 135 Amoebiasis 3 1 ko05146 136 mRNA surveillance pathway 3 2 2 ko03015 137 Caprolactam degradation 3 1 1 1 ko00930 138 Carbohydrate digestion and absorption 3 2 2 ko04973 139 Ether lipid metabolism 3 1 1 1 ko00565 140 Toluene degradation 3 1 ko00623 141 Shigellosis 3 1 2 2 ko05131 142 Tight junction 3 2 1 ko04530 143 Phototransduction - fly 3 2 2 ko04745 144 Vitamin B6 metabolism 3 1 1 ko00750 145 Drug metabolism - other enzymes 2 ko00983 146 Leukocyte transendothelial migration 2 2 1 ko04670 147 Leishmaniasis 2 1 1 ko05140 148 D-Glutamine and D-glutamate metabolism 2 ko00471 149 Phosphatidylinositol signaling system 2 1 ko04070 150 Cell cycle - Caulobacter 2 1 1 1 ko04112 151 Naphthalene degradation 2 1 1 ko00626 152 Bacterial secretion system 2 1 ko03070 153 Ethylbenzene degradation 2 1 ko00642 154 C5-Branched dibasic acid metabolism 2 ko00660 155 Base excision repair 2 1 1 ko03410 156 Fc gamma R-mediated phagocytosis 2 2 1 2 ko04666 157 Viral myocarditis 2 2 1 ko05416 158 GnRH signaling pathway 2 1 1 2 ko04912 159 Flavone and flavonol biosynthesis 2 2 2 ko00944 160 Arrhythmogenic right ventricular cardiomyopathy (ARVC) 2 2 1 ko05412 161 Dilated cardiomyopathy 2 2 1 ko05414 162 Apoptosis 2 1 1 ko04210 163 ECM-receptor interaction 2 1 ko04512 164 Hypertrophic cardiomyopathy (HCM) 2 2 1 ko05410 165 DDT degradation 2 2 1 ko00351 166 Ubiquitin mediated proteolysis 2 1 1 ko04120 167 Adherens junction 2 2 1 ko04520 168 Melanogenesis 1 1 ko04916 169 Vascular smooth muscle contraction 1 1 ko04270 170 Basal transcription factors 1 1 1 ko03022 171 Lipopolysaccharide biosynthesis 1 ko00540 172 Nucleotide excision repair 1 ko03420 173 Stilbenoid, diarylheptanoid and gingerol biosynthesis 1 1 1 ko00945 174 Steroid biosynthesis 1 ko00100 175 RIG-I-like receptor signaling pathway 1 ko04622 176 Cardiac muscle contraction 1 1 1 ko04260 177 Phototransduction 1 1 ko04744 178 Sulfur relay system 1 1 1 ko04122 179 Gastric acid secretion 1 1 ko04971 180 Retinol metabolism 1 1 1 ko00830 181 Circadian rhythm - plant 1 ko04712 182 Mismatch repair 1 ko03430 183 Salivary secretion 1 1 ko04970 184 Benzoxazinoid biosynthesis 1 ko00402 185 Fatty acid elongation in mitochondria 1 ko00062 186 Notch signaling pathway 1 1 1 1 ko04330 187 Thiamine metabolism 1 1 1 ko00730 188 DNA replication 1 ko03030 189 Indole alkaloid biosynthesis 1 ko00901 190 Synthesis and degradation of ketone bodies 1 1 1 ko00072 191 Long-term potentiation 1 1 ko04720 192 Diterpenoid biosynthesis 1 ko00904 193 SNARE interactions in vesicular transport 1 ko04130 194 Glioma 1 1 ko05214 195 Olfactory transduction 1 1 ko04740 Expression profile of differential abundance protein species
-
The differential abundance protein species in the three different comparisons were sorted on the basis of the abundance patterns during different CA stages (Fig. 2). The patterns of differential abundance protein species varied widely and were clustered roughly into 11 groups. Among these clusters, cluster C only contained three protein species, namely pertin acetylesterase family protein, macrophage migration inhibitory factor family protein and chloroplast nucleoid DNA binding protein. These protein species were down-accumulated in three comparisons, especially in DA vs NA and DA vs CA. Furthermore, the cluster G and H were formed only by FERONIA receptor-like kinase and ribosomal protein respectively. They were dramatically down-accumulated and up-accumulated respectively in DA vs NA and CA vs NA. However, their abundance had very small change in DA vs CA. Except above minor clusters, the other eight clusters constituted at least six protein species. The biological functions of the protein species involved in these large clusters were classified based on GO enrichment analysis (Supplemental Table S5). Interestingly, most of differential abundance protein species functioned in or composed cell part, cell, organelle, catalytic activity, metabolic process, binding, cellular process, organelle part and response to stimulus. Cluster A and cluster D were formed by a small number of protein species. The protein species in cluster A were up-accumulated in DA vs NA and DA vs CA, were down-accumulated in CA vs NA, while the protein species in cluster D had an opposite profile. Similarly, cluster F and I also had an opposite abundance profile. The protein species abundances in cluster F were increased in DA vs CA and decreased in both CA vs NA and DA vs NA. Cluster B and E were respectively consisted of 36 and 49 protein species that showed reduced abundance in three comparisons, especially for the protein species in cluster E among the comparisons of CA vs NA and DA vs NA. Cluster J and neighboring cluster K had similar abundance profiles because the protein species involved in these clusters were increased at different levels among the three comparisons.
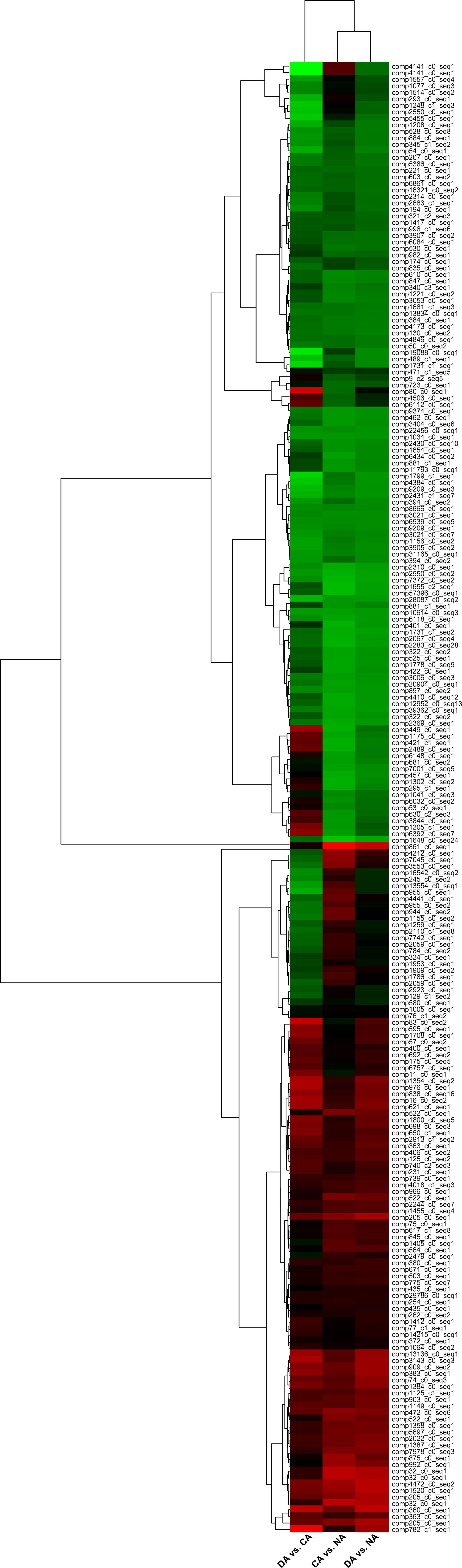
Figure 2.
Hierachical clustering of proteins showing different abundance profiles across different samples. The data were transferred using log2.
Among these differential abundance protein species, some stress-related or responding protein species were identified. These protein species included ribosomal proteins (RPs), photosynthesis related proteins, energy metabolism proteins, osmosensing-responsiveness proteins, antioxidation-related proteins, and some signal transduction, transporter and post-translationally modified proteins, etc. These protein species were grouped into different clusters. Most of these protein species were up-accumulated in CA and/or DA stage compared to NA, indicated that complex proteomics changes were happened during CA procedure in tea plant.
Analysis of the association between proteome and transcriptome
-
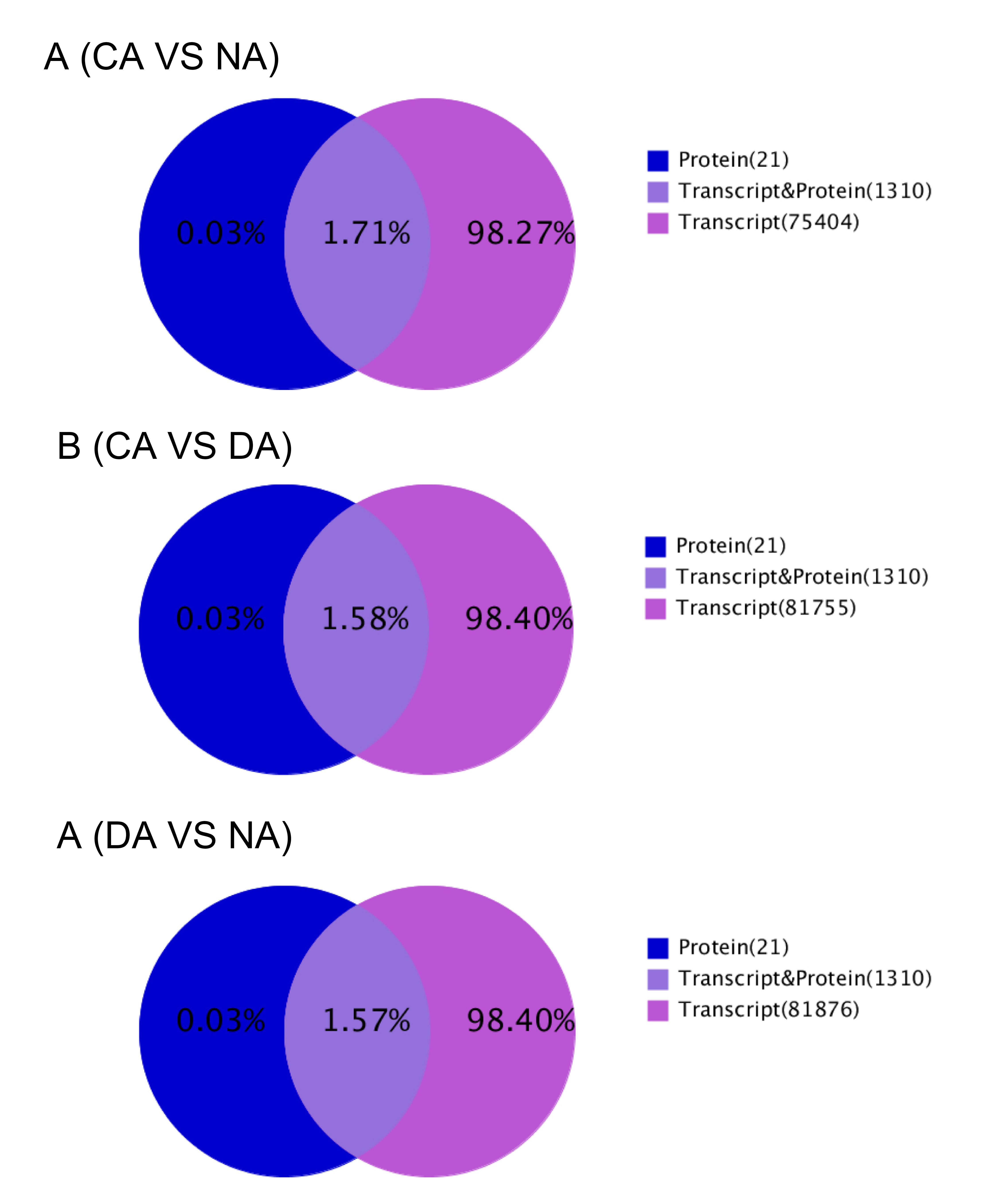
To illuminate the potential correlation between proteome and the corresponding transcriptome, all the identified proteins were correlated to corresponding transcriptome first, and thereafter association analyses were carried out between identified proteins and corresponding differentially expressed genes among the comparisons between different CA stages. Results showed that 1310 proteins out of total 1331 identified proteins were successfully associated with transcripts (Supplemental Fig. S3). Unexpectedly, the association analysis results showed that the identified proteins had low correlation coefficients (r) with the cognate genes level in the comparisons of CA vs NA, DA vs CA and DA vs NA in transcriptome analyses, with r values of 0.0023, 0.1519 and 0.1120 respectively (Fig. 3). Because the value of correlation coefficient was close to zero, the protein level was poorly correlated with transcript levels. To further show the details about the expression patterns of identified proteins and its corresponding associated gene, clustering analyses of expression patterns was implemented (Supplemental Fig. S4). The clustering results showed that the identified protein species and differentially expressed genes were mainly grouped into three kinds of cluster. First is positive correlation, such as the cluster F in CA vs NA, cluster C in CA vs DA and cluster D in DA vs NA. Second is confused correlation, which includes both week negative and positive correlation, such as cluster G in CA vs NA, cluster D in DA vs CA and cluster E in DA vs NA. These correlation relationships took a large share. Third is clear negative correlation, such as the cluster B, C, D, E and I in CA vs NA, cluster E in DA vs CA, cluster C, F, G in DA vs NA. Main proteins included in these groups have functions that include beta-primeverosidase, glycyl-tRNA synthetase, alpha-glucan water dikinase, AT-HF, phosphoenolpyruvate carboxylase, carbonic anhydrase, hydrolase family protein and plastocyanin-like domain-containing protein.
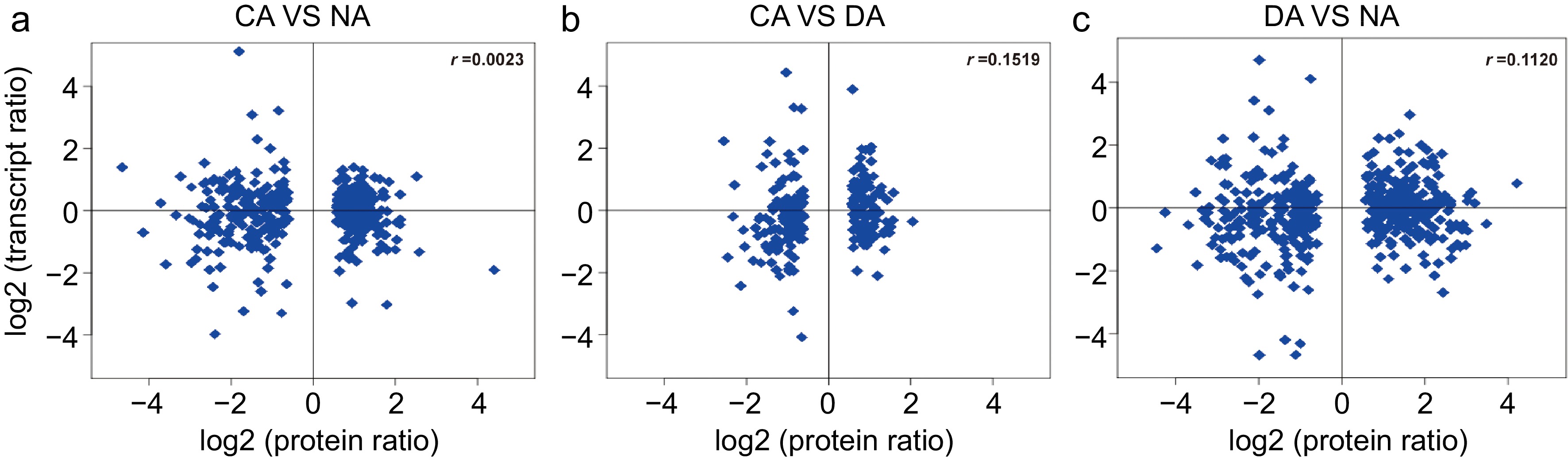
Figure 3.
Correlation analysis of transcript (log2 FPKM value) and protein (log2 iTRAQ value) among different samples. (a) CA vs NA; (b) CA vs DA; (c) DA vs NA.
Transcriptional expression analysis by qRT-PCR
-
In order to validate the correlationship between proteome and the corresponding transcriptome, 20 protein species were chosen for qRT-PCR analysis, including 17 differential abundance protein species and three unchanged abundance protein species. The expression pattern analysis showed that only seven genes had similar patterns with iTRAQ results (Fig. 4). The results were consistent with the association analysis results between proteome and transcriptome. These results may be due to various post translational modifications and other complex regulatory networks in tea plant response to cold stress.
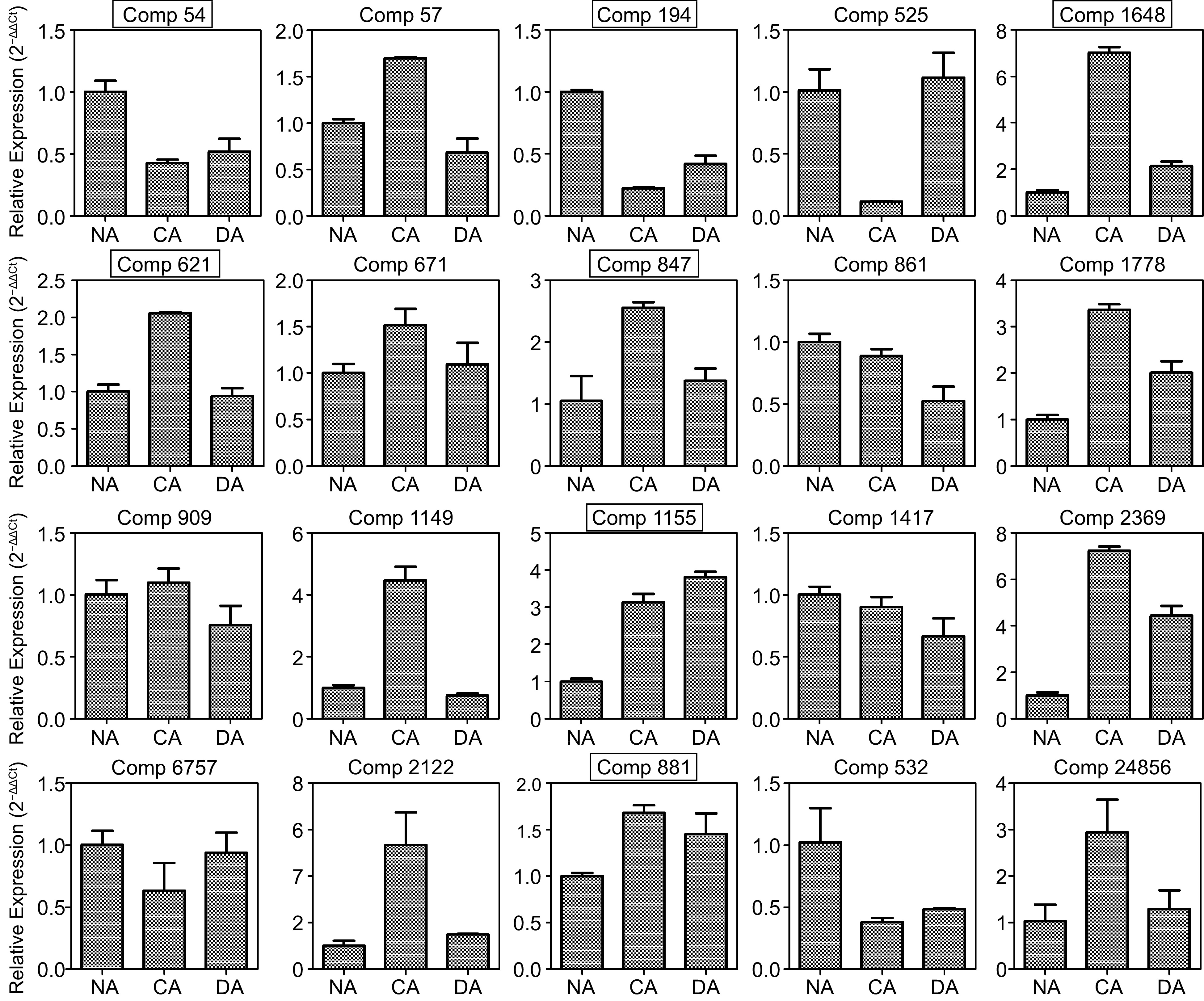
Figure 4.
Analysis of transcript levels of the selected proteins among NA, CA and DA stages by qRT-PCR. Those genes which showed similar patterns are marked using black boxes. All data are the mean ± SD (n = 3).
-
As a perennial, evergreen and originating from tropical regions, low temperature is widely accepted as the most critical factor limiting tea plant growth and geographical distribution. An understanding of the adaptive mechanisms under low temperature stress of tea plant is necessary to enhance its cold tolerance. Although some studies have focused on the cellular, physiological, metabolic and transcrioptomics changes in tea plant during CA procedures[4, 13, 14, 16, 28−30], but the molecular mechanism at the proteome level remains unclear. In the present study, we conducted comparative proteomic analysis and iTRAQ labeling method to examine the whole protein profile changes at the different CA states in tea plant leaves. A large number of differential abundance proteins, putatively related to cold stress, were identified. Comparisons of the differentially accumulated proteins revealed that more differentially accumulated proteins were detected among the comparisons of CA vs NA and DA vs NA. The result indicates that many changes which resulted from CA remained unchanged in DA. This may indicate that de-acclimated plants might have greater cold resistance than NA plants, and might be more amenable to re-acclimation than un-acclimated plants. A similar response was observed in Arabidopsis with regards to transcript changes of those genes involved in photosynthesis, calcium signaling and general stress responses maintaining acclimated expression patterns[31]. However, more work needed to be done to test this hypothesis. GO enrichment analysis is a commonly used tool to determine the potential function of differentially accumulated proteins in different data set comparisons[23]. In this study, the significant different GO terms among different comparisons between NA, CA and DA were obtained using GO enrichment analysis. As has been observed previously in other systems[21, 32−35], our results showed that the differentially accumulated protein species during CA and DA mainly involve in cell wall, photosynthesis, energy, protein synthesis, metabolism, antioxidation, carbohydrate metabolic process, and binding. Recent studies showed that these biological processes or cellular components were common to CA. Degand et al. reported that the differentially accumulated proteins identified from chicory root after CA were mainly classified into the functional categories of protein synthesis, metabolism, energy or cell structure[36]. Kosmala et al. found that proteins related to photosynthetic machinery, cell energy and cell metabolic pathways played important roles in the CA procedure of Festuca pratensis[32]. The proteomic analysis of Thellungiella rosette leaves under cold stress revealed that most identified proteins were involved in photosynthesis, defense response, cell wall and cytoskeleton, RNA metabolism, energy pathway, protein metabolism and signal transduction pathways[37]. The proteomic studies of the CA mechanism in sunflower found that those cold-responsive proteins from three different cold tolerant lines were mostly involved in metabolism, protein synthesis, energy, and defense processes[10]. Moreover, the proteomic results in plantain also indicated that the majority of differentially accumulated proteins were involved in oxidation-reduction, photosynthesis, and several primary metabolic processes[21]. Our results also showed that 20 pathways were significantly enriched in tea plant during CA process at transcriptome level, and the metabolism was the largest category, which included 'carbohydrate metabolism pathway', 'energy metabolism pathway', 'xenobiotics biodegradation and metabolism' and 'lipid metabolism', etc[4]. Therefore, it can be assumed that the CA of tea plant is characteristic of many previously identified integral metabolic changes and suggest that many of the previous regulatory mechanisms controlling CA and DA can be used to direct future research into improving cold tolerance of tea plant.
Interestingly, according to the significantly different GO terms listed in Table 1, cell wall is a significantly over-represented ontology that is specifically associated with DA. Cell wall is the first physical barrier and it plays a vital role in plant responses to abiotic stress[38]. Previous research suggested that many changes happen in the cell wall when plants were placed under cold stress, such as the increase in weight of cell walls, cell wall composition changes, and expression of cell wall-related gene changes and those changes showed close relationships with plant cold resistance[39−41]. Our results were consistant with previous studies and provided some novel findings for tea plant CA mechanism research.
Tea plant is an evergreen woody plant, and thus adjusting photosynthetic processes to deal with alterations in membrane fluidity and structure are important[37, 42]. We observed the terms of plastid part, thylakoid light-harvesting complex, chloroplast thylakoid membrane, light-harvesting complex, plastid thylakoid membrane, NADP metabolic process and DANPH regeneration being over-represented among differentially accumulating proteins in DA vs CA, plastid envelope in DA vs NA, plastid stroma in both CA vs NA and DA vs CA, and photosynthetic electron transport chain and photosynthesis in both DA vs CA and DA vs NA in concurrence with the hypothesis that modifications in photosynthesis are required for CA and DA in tea plant. These observations are consistent with the fact that photosynthetic processes can lead to greater production of damaging oxidative radicles when thylakoid membranes undergo changes of state during chilling[43]. Furthermore, reactive oxygen species scavenging is an important mechanism needed for coping with oxidative stress under cold stress in plants[44]. Consequently, over-representation of the terms of oxidation reduction and oxidoreduction coenzyme metabolic process suggest involvement of antioxidative mechanism. The terms of electron transport chain was significantly over-represented in both DA vs CA and DA vs NA, while in DA vs CA was energy reserve metabolic process. Moreover, the alterations in protein synthesis were indicated by over-representation of the terms of ammonia ligase activity, acid-ammonia (or amide) ligase activity and ribosome biogenesis in both DA vs CA and DA vs NA comparisons. Energy production related proteins were significantly up-regulated in rice leaf blades and were down-regulated in poplar leaves under a chilling environment[45, 46]. The terms of metabolic process and other terms involved in metabolism were over-represented among protein annotations in DA vs NA. The terms of carbohydrate metabolic process in CA vs NA and racemase and epimerase activity, acting on carbohydrates and derivatives in DA vs CA are involved in carbohydrate metabolism. The term of binding has significant difference in both CA vs NA and DA vs NA. Significant different terms of membrane part and fatty acid process in DA vs NA were also detected. Membrane modifications – particularly regarding alterations in fatty acid composition, have long been associated with CA processes[47]. Likewise, it is well known that carbohydrate accumulation can be protective against cold stress[48]. And these results were consistent with our previous study at the transcriptome level[4].
CA is an important mechanism for perennial plants to obtain or enhance freezing tolerance[1]. Extended freezing temperatures in winter pose a great challenge for the survival of evergreen perennials such as tea plant, and, along with dormancy formation, such plants develop physiological and molecular changes to successfully archieve overwintering[49]. Because freezing of extra-cellular water pose significant challenges in regards to dehydration, cellular changes in water content, water status and osmotic potentials are essential events[50]. Consequently, cells decrease water content during fall or early winter and accumulate osmoprotectants such as specific storage proteins, sugars, starch and alter membrane chemistries to better deal with dehydration[51]. These adaptive mechanisms rely in part on gene induction and regulation, resulting in related protein enrichment in the fully acclimated stage. Therefore, the functional category analysis revealed that cell wall, photosynthesis, energy, protein synthesis, metabolism, antioxidation, carbohydrate metabolic process, and binding take crucial roles in tea plant CA.
Pathway enrichment analysis was conducted to determine the main signal transduction and biochemical metabolic pathways involved by those differentially accumulated proteins in a previous study. The results showed that microbial metabolism in diverse environments, metabolic pathways and biosynthesis of secondary metabolites were the top three enriched pathways. In concordance with GO analyses above, carbon fixation in photosynthetic organisms, ribosome, carbon fixation in photosynthetic organisms, starch and sucrose metabolism, protein processing in endoplasmic reticulum, plant-pathogen interaction, oxidative phosphorylation and photosynthesis also take a large proportion of the differentially accumulated proteins. The changes of general metabolism and photosynthesis are the main responses of tea plant to low temperature. Accumulation of secondary metabolic products is an important character for tea plant. Many of the differentially accumulating proteins are involved in secondary metabolism related pathways. The role of these secondary metabolites in CA and DA are difficult to envision. For example, we observed increases in gallic acid and its derivatives which are known to have anti-oxidant properties, and thus might have protective roles in freezing stress[48]. However, increases in gallic acid production are not commonly observed during CA processes of other non-evergreen systems. It is well noted in the literature that protein content in plant cell can change within hours of low temperature exposure, and that protein metabolism play important roles in CA and freezing stress tolerance[52]. The enrichment of ribosome and protein processing in endoplasmic reticulum in this study also indicates the close correlation between protein synthesis and CA. Interestingly, a large number of proteins were involved in the pathways of microbial metabolism in diverse environments and plant-pathogen interaction. This was consistent with the results found in the plasma membrane of oat and rye after CA[53]. Recent studies report that some of the pathogenesis-related proteins are induced during winter months and have been shown to have antifreeze activity, cryoprotective activity, or antifungal activity[49].
Furthermore, the signal transduction pathway plays a pivotal role in the response to the stress of low temperatures[54]. Plasma membrane play an important role in response to low temperature stress[47]. Plasma membrane can sense and transduce cold signals and then signal responses that alter its structure, chemical composition and function to improve cold resistance[11, 53, 55]. Li et al.[56] and Takahashi et al.[53] had identified hundreds of differentially abundant plasma membrane proteins in Arabidopsis, oat and rye using shotgun proteomic technology suggesting that these proteins may have a role in CA and freezing tolerance development. These proteins included signal transduction, disease/defense-related, energy-related, transporter and post-translationally modified proteins, etc. Phosphatidic acid is one of the major membranous second-messenger molecules and is produced by phospholipase D. During CA, plants generally increase their phospholipase D levels and phosphatidic acid content which is correlated with enhanced low temperature resistance[8,57]. Heat shock proteins are a kind of membrane protein that act as molecular chaperones[58], and usually play crucial roles in response to cold stress by re-establishing normal protein conformation and thus cellular homeostasis[53,59]. In tea plant, we also found phospholipase D (comp1149_c0_seq1) and heat shock proteins (comp671_c0_seq1, comp1412_c0_seq1 and comp16542_c0_seq2) were up-accumulated in CA and DA compared with NA. The plasma membrane contains many proteins, and some of those involved in cold-responsive process in tea plant were also reported in other plants[53,56]. These include, but are not limited to, proteins such as early response to dehydration proteins (ERDs), fasciclin-like arabinogalactan proteins (FLAs), aquaporins, ATPases, clathrins weren't differentially accumulated in our results (Supplemental Table S1). Studies using isolated plasma membrane should be conducted to elucidate the accurate changes of plasma membrane-specific proteins during CA in the future.
-
In the present study, 1,331 proteins were identified from NA, CA and DA tea leaves using iTRAQ analysis. 407 and 477 proteins were differently accumulated in comparison NA vs CA and DA Vs CA respectively. Function and KEGG pathway analysis revealed that those differently accumulated proteins were mainly mapped onto the metabolic, biosynthesis of secondary metabolites, microbial metabolism in diverse environment, ribosome, sugar metabolism, protein processing, photosynthesis and plant-pathogen interaction pathways. Further GO enrichment analysis indicated that those proteins were mainly involved in protein synthesis, photosynthesis, energy, sugar metabolism, antioxidation and stress defense. Correlation analysis showed that the proteome changes were not well-correlated with corresponding gene transcription changes. Overall, our study revealed general information about the proteome changes in tea plant leaf during NA, CA and DA procedures and provided some new insights on cold tolerance mechanism in tea plant.
This work was supported by the National Natural Science Foundation of China (U22A20499), the China Agriculture Research System of MOF and MARA (CARS-19), the Chinese Academy of Agricultural Sciences through an Innovation Project for Agricultural Sciences and Technology (CAAS-ASTIP-2021-TRICAAS) and the special project of Zhejiang province (2020R52036).
-
The authors declare that they have no conflict of interest. Xinchao Wang is the Editorial Board member of Beverage Plant Research who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board members and his research groups.
-
# These authors contributed equally: Changqing Ding, Xinyuan Hao
- Supplemental Fig. S1 Experimental design and wokeflow of the iTRAQ analysis on tea plant during different cold acclimation stages.
- Supplemental Fig. S2 Protein abundance distribution between the three different sample stages (CA vs NA, CA vs DA and DA vs NA).
- Supplemental Fig. S3 Venn charts for correlation between proteome and transcriptome database.
- Supplemental Fig. S4 Clustering analyses of expression patterns between identified proteins and its corresponding associated gene (A. CA vs NA; B. DA vs CA; C. DA vs NA).
- Supplemental Table S1 Primers used for quantitative RT-PCR.
- Supplemental Table S2 Raw determination data in proteome analysis (sheet "raw determination data"), raw data of proteomic accumalation analyses comparing with transcriptome data (sheet "expression data analysis"), and KEGG and GO term annotation for detected proteins (sheet "KEGG and GO term annotation").
- Supplemental Table S3 Total pathway analysis results of total and enriched protein species in the comparisons among different samples.
- Supplemental Table S4 Information of the total identified and differentially accumulated protein species mapped in KEGG pathway.
- Supplemental Table S5 Differentially accumulated protein species among the three comparisons (CA vs NA, DA vs NA and DA vs CA) (sheet "differentially accumulated proteins") and Gene Ontology (GO) enrichment analysis on the basis of clustering analysis (sheet "GO analyses of large clusters").
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Ding C, Hao X, Wang L, Li N, Huang J, et al. 2023. iTRAQ-based quantitative proteomic analysis of tea plant (Camellia sinensis (L.) O. Kuntze) during cold acclimation and de-acclimation procedures. Beverage Plant Research 3:16 doi: 10.48130/BPR-2023-0016 

iTRAQ-based quantitative proteomic analysis of tea plant (Camellia sinensis (L.) O. Kuntze) during cold acclimation and de-acclimation procedures
- Received: 24 March 2023
- Revised: 13 June 2023
- Accepted: 19 June 2023
- Published online: 14 July 2023
Abstract: To gain a better understanding on the mechanism of cold acclimation in tea plant [Camellia sinensis (L.) O. Kuntze] at the proteome level, an iTRAQ based quantitative proteome analysis was carried out to identify differentially accumulated proteins in the mature leaves which were collected at non-acclimated (NA), fully acclimated (CA) and de-acclimated (DA) stages. 407 and 477 proteins identified from CA and DA showed significant abundance changes (at 95% confidence) compared with NA, respectively. Moreover, 251 protein species changed their abundance in DA compared with CA. Those differential abundance protein species were mainly involved in metabolism, cell wall, photosynthesis, energy, protein synthesis, antioxidation, carbohydrate metabolic process and binding, and mapped to the pathways of biosynthesis of secondary metabolites, microbial metabolism in diverse environment, ribosome, metabolic pathway, sugar metabolism, protein processing, photosynthesis, and plant-pathogen interaction pathway. However, no significant correlation was detected between the identified proteins and cognate gene transcript levels by correlation analysis and qRT-PCR analysis. This study presents a comprehensive proteome in mature leaves at different cold acclimation status and provides new insights into cold acclimation mechanisms in tea plants.
-
Key words:
- Tea plant (Camellia sinensis) /
- Cold acclimation /
- iTRAQ /
- Proteome /
- Cold-responsive protein
{kind=link}
{kind=link}
{kind=link}
{kind=link}