









-
Ecological interactions are evolutionarily conserved, indicating a tendency of closely related species to interact with similar partners[1]. Thus, phylogenies can significantly contribute to the understanding of interactions, even across trophic levels[2,3]. In particular, plant phylogeny leaves an imprint in root-associated microbial communities[4−8]. The magnitude of the plant's phylogenetic signature belowground is especially strong on host-dependent microbial partners based on their history of coevolution[3]. Ultimately, such a phylogenetically structured top-down control can further affect the rates of essential ecosystem processes[9].
Arbuscular mycorrhizal (AM) fungi form obligate symbioses with the roots of most land plants[10]. The AM symbiosis is arguably the world's most prevalent and ancient mutualism known, traced back to early land colonization by plants[11]. Local host preference is frequently reported as a factor structuring AM fungal communities[12,13], as closely related woody plants tend to interact with closely related AM fungi[14]. The woody plant–AM fungi networks are highly interconnected and explained by plant and AMF phylogenies. This suggests that the phylogenetic niche conservatism in woody plants and their AM fungal symbionts could contribute to interdependent AM fungi and plant community assembly. Also on a local scale, the phylogenetic composition of plant communities, as well as plant life history traits, have been shown to impact the phylogenetic structure of AM fungal assemblages[15,16]. For example, the effect of plant life history traits was depicted as that in the order annual herbs–perennial herbs–semi-woody plant species, there was a transition from phylogenetic clustered to over-dispersed AM fungal communities. Most of the AM fungi preferentially associated with annual plants belonged to Glomeraceae, showing a phylogenetic clustering pattern. However, conflicting results have been reported on a broader scale. At the global scale, variations in AM fungal communities are similar to the levels of variations seen in plant families[17], as plants within the same family may have inherited similar traits or may share similar ecological niches that select for particular AM fungal communities. A global meta-analysis however indicates that plant-fungal association patterns are poorly influenced by host phylogeny[18]. Further, there are few studies about whether AM fungi-host plant preference could exert selection within AM fungal communities.
In addition to host plant effects, environmental filtering (e.g., soil physical and chemical properties) and geographical dispersal are also capable of affecting AM fungal communities. However, the relative importance of AM fungi-host preference, environmental filtering and geographical dispersal has yet to be tested to check whether AM fungi-host preference is a central mechanism structuring AM fungal communities. In the present study, a sampling of the roots of plant individuals across the Xilingol steppe in northern China were manipulated. Whether the phylogenetic divergence of host plants may be a main driver for variations of AM fungal communities at the regional scale was tested, at which spatial and environmental drivers can concurrently play a relevant role.
-
The sampling area represents a typical steppe in the Xilingol Grassland in northern China, which is located in the mid-latitude inland area with a semi-arid climate and covers a total area of 179,600 km2 (Fig. 1; Supplementary Table S1). Ten sites were randomly chosen across the sampling area in August 2022 (Fig. 1; Supplementary Table S1). To collect roots, soil was excavated to a depth of 0.4 m because this is where fine roots are most abundant. Then roots that were connected to shoots were identified, and one plant individual was randomly chosen and its root samples were collected manually with sterile gloves (sprayed with 70% EtOH) put in plastic bags, and refrigerated. Root samples were transported to the laboratory, washed free of soil, and stored for molecular analysis. The location of each sampled individual was recorded in GPS. At each of these sampling sites, 29–30 plant individuals were sampled. Only adult plant individuals were chosen and seedlings and juveniles were not included. A total of 296 root samples (plant individuals) were collected, encompassing 76 plant species from 29 plant families (see the phylogenetic tree in Supplementary Fig. S1).
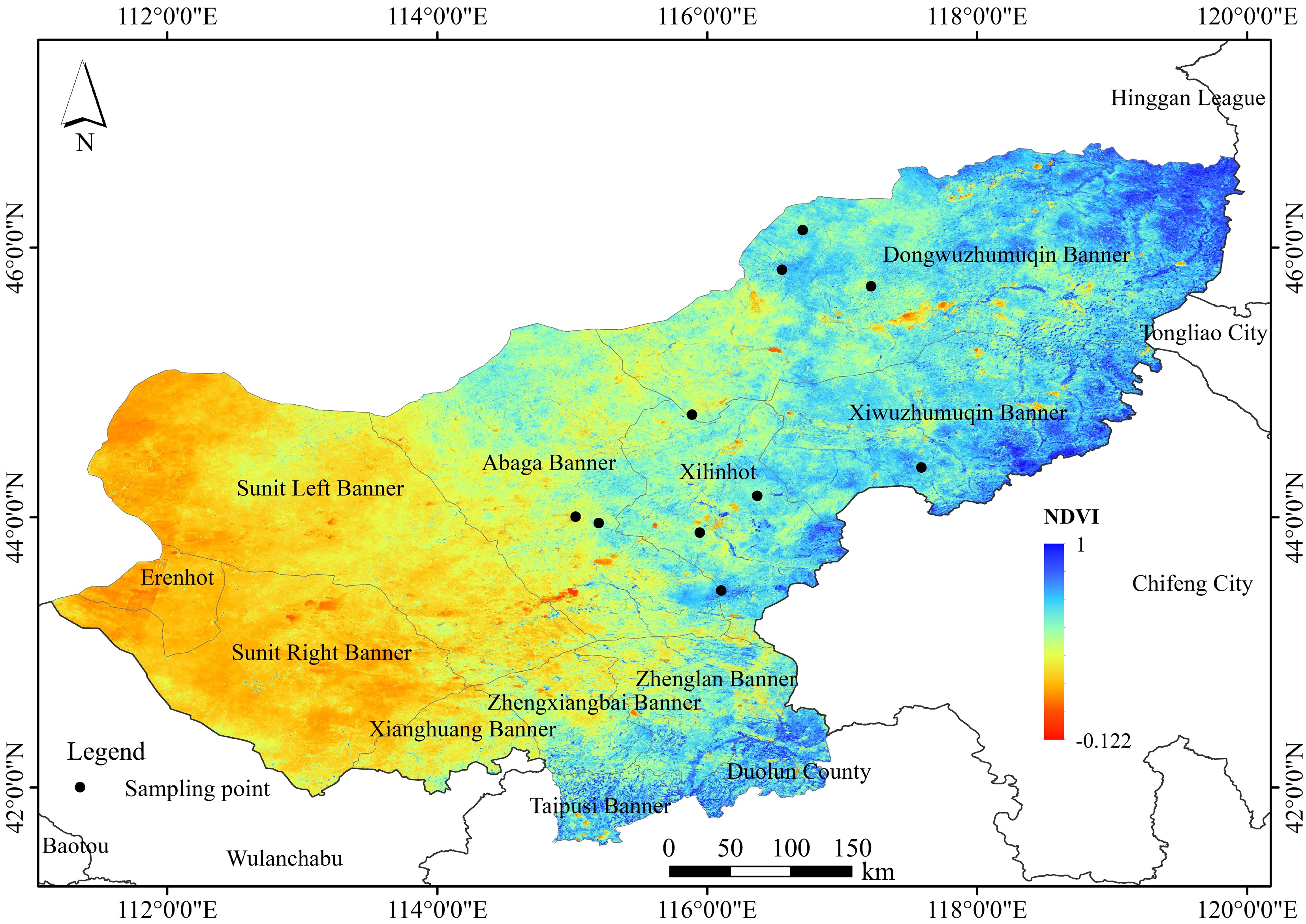
Figure 1.
Geographic distribution of the 10 sampling sites containing 296 sampling individuals. Sampling sites are described in detail in Supplementary Table S1.
For each sampled plant individual, the longitude, latitude, and altitude were recorded with an eXplorist 210 GPS (Magellan, San Dimas, CA, USA). Mean annual temperature (MAT) and mean annual precipitation (MAP) were compiled from the National Meteorological Bureau of China database. Data were compiled by interpolating data of daily mean temperature and daily precipitation records (1961–2016) from 716 climate stations across China (
http://data.cma.cn ). Rhizosphere soil was collected by gently shaking off the soil adhering to the roots for measuring soil characteristics. For each root sample, there is a corresponding rhizosphere soil sample, leading to a total of 296 rhizosphere soil samples.Molecular analysis of AM fungal communities
-
To remove AM fungal spores or hyphae from the root surface, roots were sonicated at low frequency for 3 min (30-s bursts followed by 30-s rests performed three times). Genomic DNA was extracted from a 0.2 g (dry) subsample of fine roots with the FastDNA SPIN Kit (MP Biomedicals, Santa Ana, CA, USA) following the manufacturer's protocol. The extracted DNA was dissolved in 50 μL TE buffer, quantified by spectrophotometer, and then stored at −20 °C before further use. The root AM fungal community was described using the Illumina Miseq platform with the primer set AMV4.5NF (5′-AAGCTCGTAGTTGAATTTCG-3′) / AMDGR (5′-CCCAACTATCCCTATTAATCAT-3′) which targets the 18S SSU rRNA gene region[19]. PCR was performed with 35 cycles (95 °C for 45 s, 58 °C for 45 s, and 72 °C for 1 min) and a final extension at 72 °C for 7 min in 50-μL reaction mixtures (1.25 mM deoxynucleoside triphosphate, 2 U Taq DNA polymerase (TaKaRa, Shiga, Japan), 15 μM AMF primers, and 50 ng genomic DNA). PCR products were purified using a QIAquick PCR Purification kit (QIAGEN), quantified using a NanoDrop ND-1000 instrument (Thermo Scientific, USA) (confirmed using the ratio of A260/280 between 1.8–2.0), and were normalized in equimolar amounts before sequencing. Sequencing reads were assigned to samples, and the corresponding paired reads were merged if the overlap was 100% identical using FLASH (V1.2.7,
http://ccb.jhu.edu/software/FLASH/ ). The reads were quality-filtered with QIIME 1.6.0[20]. Default settings for Illumina processing in QIIME were used [minimum number of consecutive high-quality base > 75% total read length, maximum number of consecutive low-quality base = 3, last quality score considered low-quality = 3, maximum number of ambiguous (N) characters = 0] as recommended by Bokulich and colleagues[21]. After removing chimera sequences with UCHIME (Version 4.2)[22], OTU (operational taxonomic units) classification at 0.97 similarity and taxonomic assignment were acquired by blasting against the MaarjAM database (http://maarjam.botany.ut.ee )[23] following the criteria of Davison et al.[24]. After removing singletons, the longest sequence for each OTU was chosen as the representative sequence. The representative sequences were aligned with ClustalW[25]. Because an even sequencing depth per sample is required for beta diversity calculations, the OTU table was rarefied to 502 sequences per sample. There were 188 and 167 OTUs in the OTU table before and after the rarefaction, respectively. The maximum likelihood phylogenetic trees of AM fungi were inferred with RAxML v7.0.3[26] using the GTRCAT model and 1,000 bootstrap replicates. Six independent phylogenetic trees were constructed based on different seeds. Phylogenetic trees of AM fungi are provided as Supplementary Fig. S1.Plant phylogeny
-
The phylogenetic relationships between the 76 studied plant species was resolved by firstly matching the species names to the tips of the megaphylogeny of vascular plants (i.e., GBOTB.extended.TPL) embedded within the R software package V.PhyloMaker2[27]. A total of 54 of the 76 species names were matched. Secondly, the remaining 22 unmatched species were bound to the megaphylogeny by adopting the 'S3' scenario of the function 'phylo.maker' in V.PhyloMaker2. Thirdly, the expanded megaphylogeny was trimmed using the 'drop.tip' function in the ape package for R[28], to retain the 76 study species only (Supplementary Fig. S1).
Soil chemical characteristics
-
The characteristics of the rhizospheric soil samples were assessed by determining their total nitrogen (TN), extractable phosphorus, pH, and soil organic carbon (SOC). Soil TN was determined by performing elemental analyses (Elementar Vario MACRO, Germany). Extractable P concentrations were determined using a spectrophotometer (UV-1600 spectrophotometer, Beijing). The standard Walkley-Black potassium dichromate oxidation method was employed for obtaining SOC. Then, a 1:1 ratio of soil to water slurries was used to measure soil pH with an acidometer (HANNA, Padova, Italy).
Phylogenetic diversity metrics
-
Several indices of phylogenetic community structure and turnover were calculated. First, the phylogenetic structure of AM fungal communities was described by calculating a matrix (matrix P) that contains the composition of species fuzzy‐weighed by their pairwise phylogenetic similarities[29] with the R package PCPS[30]. In matrix P, each OTU has a value per sample that increases as the phylogenetic distance between neighbouring OTUs decreases. Ordination techniques allow reducing matrix P to represent the phylogenetic structure at the sample level. Principal coordinate analysis was performed with Euclidean distances and extracted the sample scores along the first axis, which represents the principal component phylogenetic structure (PCPS1). This axis captures the deepest phylogenetic divergences among lineages[31,32].
Second, the phylogenetic turnover of AM fungal communities from each plant individual was computed as betaMNTD (between-assemblage analogs of mean nearest taxon distance) and betaMPD (between-assemblage analogs of mean pairwise distance) as described by Fine & Kembel[33]. BetaMNTD is the mean nearest taxon distance between pairs of species drawn from two distinct communities and is sensitive to the changes of lineages close to the phylogenetic tips, while betaMPD is sensitive to tree-wide distributions of lineages. Further, betaNTI (between-assemblage analogs of the nearest taxon index) and betaNRI (between-assemblage analogs of net relatedness index) were computed as the number of standard deviations that observed betaMNTDs or betaMPDs departed from the mean of the null model of random shuffling of taxa labels across the phylogeny[33]. BetaNTI and betaNRI were computed as the number of standard deviations that observed betaMNTDs and betaMPDs departed from the mean of the null distribution, respectively. To calculate betaNTI or betaNRI, a null distribution of betaMNTDs or betaMPDs were generated by randomizing OTUs across the phylogeny (999 null iterations) based on random shuffling of OTU labels across the tips of the phylogeny. BetaNTIs and betaNRIs less than −2 indicates less than expected phylogenetic turnover (communities are more similar than expected), while betaNTIs and betaNRIs greater than +2 indicates greater than expected phylogenetic turnover. BetaNTIs and betaNRIs indicates random patterns when the values are distributed between −2 to +2 as this means distribution deviation from the null model was not significant. All four phylogenetic beta-diversity values across six replicated trees were found to be highly correlated (r > 0.90, p < 0.001) indicating reproducibility in the phylogenetic reconstructions.
Statistical analyses
-
Two approaches were used to determine the relative importance of plant phylogeny, geographic distance, and environmental variables in driving the phylogenetic community structure and turnover in AMF communities.
GLMM-MCMC approach
-
The Bayesian version of GLMM-MCMC (the generalized linear mixed model using Markov chain Monte Carlo) was performed with the six AMF phylogenetic trees with the geographic coordinates as the random factors, following the method of Stone et al.[34]. GLMMs are used for analyzing correlated non-Gaussian data, but their likelihood function is only available as a high dimensional integral, making closed-form inference and prediction impossible. Consequently, Bayesian GLMMs also have intractable posterior densities, and MCMC algorithms are typically used for conditional simulation and exploring these densities in GLMMs. In the present analysis, the three phylogenetic beta dissimilarities, PCPS1, betaMNTD, and betaMPD were used as the dependent variables in the GLMM-MCMC analysis, respectively. As betaMNTD and betaMPD were two-dimensional data in dissimilarity format, the first axis of principal coordinate ordinations was extracted and used as the dependent variable in Bayesian GLMM-MCMC analysis. Principal coordinate ordinations were also manipulated on plant phylogenetic distance and distance of environmental variable, and the first axis of each distance matrix was extracted as the independent variables. GLMM-MCMC was performed with the R package MCMCglmm[35]. The effect of predictors was estimated by calculating the 95% confidential interval of their posterior distributions[36]. 13,000 MCMC iterations were ran with a burn-in period of 3,000 iterations and default priors. Convergence of the chain was verified by the autocorrelation function of the Markov chain.
Mantel test approach
-
The Mantel test was used to explore the relationship between the phylogenetic turnover of AM fungal communities (measured as betaMNTD, betaMPD, and the distance of matrix P) and plant phylogenetic distance of host plant (betaMNTD and betaMPD of plant phylogenetic distance), geographical distance, and distance of environmental variables. As the PCPS1 is in column data format, Euclidean distance of the matrix P was used in the Mantel test. Further, partial Mantel tests were done between the distance of the matrix P/betaMNTD/ betaMPD and plant phylogeny, geographic, and environmental distance, respectively, after controlling each of the other two factors. Results were corrected for multiple testing as one series using a false discovery rate (FDR). As the phylogenetic beta-diversity values across six replicated trees showing reproducibility in the phylogenetic reconstructions, a single phylogenetic tree was used in Mantel and partial Mantel analysis. Geographic distance between samples was calculated from the latitude and longitude coordinates using the 'geosphere' packages[37]. The environmental distance was calculated as the Bray-curtis dissimilarity of environmental variables including climate (MAP and MAT) and soil physicochemical characteristics (pH, TN, extractable P, and SOC). The Mantel test examines the correlation relationship between the two matrices. In the partial Mantel test, control was implemented by calculating the correlation between the residuals of each of the two primary distance matrices after performing a linear regression on the third distance matrix.
-
A total of 167 OTUs were detected in the rarefied OTU table, among which Glomeraceae (113 OTUs) was the most dominant at the family level, followed by the Diversisporaceae (12 OTUs).
There were 43660 paired phylogenetic beta-diversities (betaMNTD or betaMPD) across the 296 samples. Calculation of the number of standard deviations that observed betaMNTDs or betaMPDs departed from the mean of the null model (i.e., betaNTI or betaNRI, respectively) showing a large proportion of both betaNTI (89.19%, 38,941 out of 43,660 paired samples) and betaNRI (93.38%, 40,770 out of 43,660 paired samples) values distributed between −2 to +2 (Fig. 2).
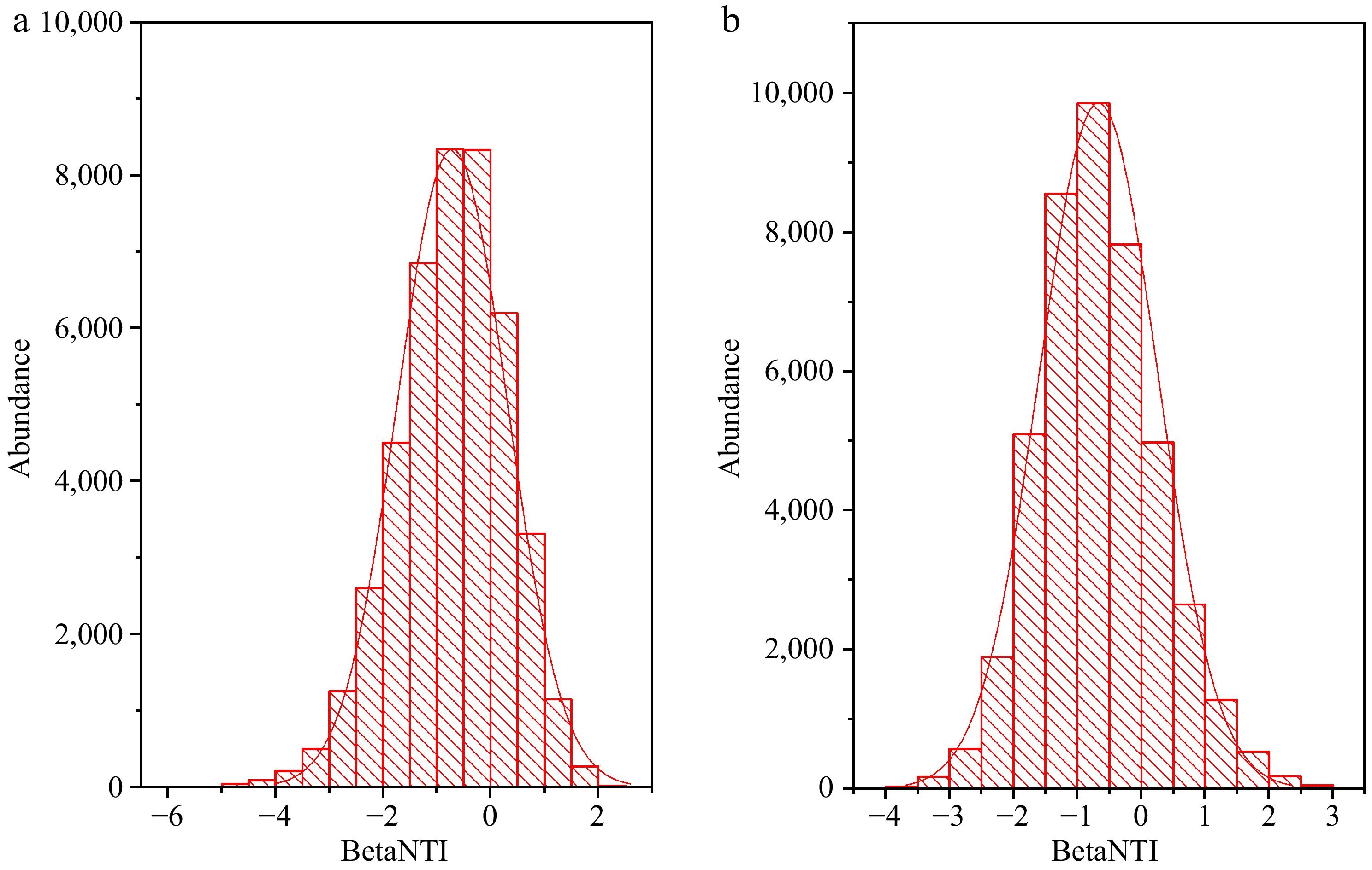
Figure 2.
Distribution of betaNTI and betaNRI values.
For the relationship between phylogenetic turnover of AM fungal communities and plant phylogeny, geographical distance, and environmental variables, the Bayesian GLMM-MCMC analysis identified geographical distance as the only significant factor in explaining phylogenetic beta-diversity of AM fungal communities (Table 1). For all the three phylogenetic beta-diversity of AM fungal communities (i.e., PCPS1, betaMNTD, and betaMPD), the 95% credible intervals of Bayesian postmean estimates explained by geographical distance did not overlap with 0 (Table 1), which suggested a significant role of geographical distance in determining beta diversity of AM fungal communities. Plant phylogeny and environmental variables were not significant in Bayesian GLMM-MCMC analysis in explaining either of the beta-diversity of AM fungal communities (Fig. 2, Table 1).
Table 1. GLMMMCMC analysis on effects of plant phylogeney, geographical distance and environmental variables on the phylogenetic turnover of AMF communities with sampling site as a random factor. Bayesian postmean estimates and 95% credible intervals are shown. Geographical distance was the only factor significantly explaining phylogenetic turnover of AMF communities, as the 95% credible interval did not overlap with 0 and p < 0.05.
PCPS1 BetaMNTD BetaMPD Post mean 95% CI Post mean 95% CI Post mean 95% CI Geographical distance 2.2 × 10−10 (1.3 × 10−12, 6.1 × 10−10) 1.9 × 10−9 (1.9 × 10−16, 8.5 × 10−9) 1.4 × 10−8 (5.0 × 10−14, 4.7 × 10−8) Plant phylogeny 4.5 × 10−3 (−1.5 × 10−2, 2.5 × 10−2) −4.6 × 10−6 (−1.1 × 10−5, 1.8 × 10−6) −7.8 × 10−6 (−2.3 × 10−5, 6.9 × 10−6) Environment 1.5 × 10−3 (−3.0 × 10−3, 5.9 × 10−3) 3.7 × 10−3 (−9.6 × 10−3, 2.0 × 10−2) −5.3 × 10−4 (−3.5 × 10−2, 3.8 × 10−2) Values in bold represent the 95% credible interval did not overlap with 0 and p < 0.05. Mantel tests showed a significant correlation between betaMNTD of AM fungal communities and plant phylogeny (FDR adjusted p < 0.05; Table 2), but the correlation between PCPS1 and betaMPD of AM fungal communities and plant phylogeny was not significant (Table 2). Geographical distance showed a significant correlation with all three phylogenetic beta diversity metrics of AM fungal communities (i.e., PCPS1, betaMNTD, and betaMPD ) (FDR adjusted p < 0.05; Table 2).
Table 2. Mantel test between unweighted betaMNTD/betaMPD of AMF communities and plant phylogeny, geographical distance, or environmental variable.
PCPS1 BetaMNTD BetaMPD r p r p r p Plant phylogeny 0.008 0.600 0.053 0.043 0.049 0.252 Geographical distance 0.051 0.004 0.179 0.003 0.058 0.012 Environment 0.002 0.998 0.021 0.037 0.022 0.924 p values were FDR adjusted. Values in bold represent the 95% credible interval did not overlap with 0 and p < 0.05. In the partial Mantel test, betaMNTD of AM fungal communities was significantly correlated with plant phylogeny after controlling for environmental variables (Table 3). When the geographic distance was accounted for in the partial Mantel test, the correlation between AM fungal communities and plant phylogeny was not significant (Table 3). The betaMNTD of AMF communities was significantly correlated with environment and geographical distance after controlling for each of the other factors (Table 3). The betaMPD of AMF communities was only significantly correlated with geographical distance after controlling plant phylogeny or environmental variables (Table 3). Correlation of betaMPD of AMF with plant phylogeny or environment was not significant after controlling geographical distance in that partial Mantel test (Table 3). The PCPS1 of AMF communities was only significantly correlated with geographical distance after controlling plant phylogeny or environmental variables (Table 3), but showed no significant correlation with plant phylogeny or environment (Table 3).
Table 3. Partial Mantel test between unweighted betaMNTD/betaMPD of AMF communities and plant phylogeny, geographical distance, or environmental variable, after controlling one of these three factors.
Controlling PCPS1 BetaMNTD BetaMPD r p r p r p Plant phylogeny Geographical distance 0.007 0.598 0.046 0.068 0.046 0.237 Environment 0.008 0.597 0.055 0.045 0.047 0.312 Geographical distance Plant phylogeny 0.051 0004 0.177 0.006 0.056 0.009 Environment 0.067 0.001 0.185 0.006 0.072 0.006 Environment Plant phylogeny 0.028 0.996 0.023 0.048 0.019 0.999 Geographical distance 0.005 0.999 0.051 0.999 0.049 0.999 p values were FDR adjusted. Values in bold represent the 95% credible interval did not overlap with 0 and p < 0.05. -
The present results showed that betaMNTD of AM fungal communities was significantly correlated with plant phylogeny, but the correlation between PCPS1 and betaMPD and plant phylogeny was not significant. The Bayesian GLMM-MCMC analysis also suggested a poor influence of plant phylogeny in explaining the phylogenetic turnover of AMF communities. The betaMNTD is the mean nearest taxon distance between pairs of species drawn from two distinct communities[33] and is sensitive to the changes in lineages close to the phylogenetic tips. The betaMPD tends to be more sensitive to the tree-wide distributions of lineages, compared to betaMNTD. PCPS1 was considered to capture the deepest phylogenetic divergences among lineages. Thus, the present results indicate that AM fungal communities colonizing plant species with close relatedness might contain taxa that are phylogenetically clustered towards the tips of phylogeny[33]. However, this correlation is reduced when controlling for geographical distance and environmental variables in the partial Mantel test, but the role of geographical distance was emphasized in both GLMM-MCMC analysis and partial Mantel analysis.
Dispersal limitation can be stochastic - when occurring through passive processes like wind - or deterministic when driven by differences in dispersal traits among taxa (or lineages)[38]. In the present study, the role of stochasticity in shaping AM fungal communities in the steppe grassland was suggested by the great importance of geographical distance, a conclusion confirmed by analyses of deviations of phylogenetic turnover from the stochastic expectation. The majority of both betaNTI and betaNRI values distributed between −2 to +2, suggesting a random pattern. This result also suggests that AM fungal communities were mainly structured by stochastic events (e.g., dispersal limitation). Plant phylogenetic distance also showed a stronger correlation with geographical distance (p < 0.001 of partial Mantel test controlling environment) than environment (p > 0.05 of partial Mantel test controlling geographical distance), indicating that stochastic events (e.g., dispersal limitation) also affected the current phylogenetic structure of plant communities. This suggested that past events generate and maintain biogeographic patterns of plants might similarly operate in the microbial world, e.g., AM fungal communities.
Dispersal limitation represents an inability of taxa to reach potentially suitable habitats in a given time frame. Nonetheless, AM fungi are found on all continents, and many approximate species-level phylogroups (phylogenetically defined groupings of taxa described by DNA sequences) have been shown to exhibit wide distributions, frequently spanning multiple continents[17,39,40]. Such broad distribution patterns suggest a highly effective long-distance dispersal strategy. However, the dispersal ability of AM fungi (e.g., dispersal of spores by wind or animals) was considered to directly influence its ability to colonize a location and therefore its geographic distribution[41−44]. Long-distance dispersal of AM fungi might be difficult since they have hypogeous and relatively large spores (0.01−1 mm) with limited dispersal ability compared to other fungal groups[45]. On this aspect, the passive dispersal of AM fungi supported the role of stochasticity in shaping AM fungal communities here. On another aspect, AM fungi disperse by large spores in the soil, mycelial fragments, and colonized root pieces[10]. Spore number, volume, and shape may be correlated with dispersal ability in fungi[46,47]. Certain functional traits of AM fungi associated with dispersal (spore number and volume) or spatial niches (e.g. allocation of hyphal biomass to soil or roots) tend to be similar among closely related species, i.e. they are phylogenetically conserved[46−48]. It might therefore be hypothesized that AM fungi with similar dispersal-related traits are more likely to co-occur than functionally dissimilar taxa. Thus, the correlation between spatial distance and both fungal and host plant phylogenetic structure suggests that distance effects on AM fungal communities may not be purely neutral, but also may reflect correlated differences in dispersal traits among lineages of the plants and fungi. The role of dispersal limitation in structuring AM fungal communities has previously been recognized on a local-[49] and global-scale[17]. Here, the analysis represents the first empirical evidence that dispersal limitation is the main determinant of AM fungal communities on a landscape scale.
Whether co-evolution has an important ongoing role in most mycorrhizal relationships is unknown[50]. AM fungi are typically considered as host generalists, although patterns of partner preference have been clearly documented[12,13,51], and plant growth responses to AM fungi depend partly on specificity between plant phylogenetic lineages and fungal taxa[52]. It has also been documented that host plant phylogeny influences the local composition of AM fungi[17]; however, tests of null models found that plant phylogeny-AM fungi relationships can be scale-dependent[13] or due to spatial effect[53]. Moreover, Põlme et al.[18] found that there was no overall evidence for a phylogenetic signal in plant-AM fungal associations on a broad scale, although the phylogenetic correlation of plant-AM fungal assemblages was detected on a local scale[15]. Here, significant correlation was found between plant phylogeny and betaMNTD of AM fungal communities in Mantel test. However, further analysis (partial Mantel test and Bayesian GLMM-MCMC) showed this AM fungi-plant relationship seems to be attributed to the similar effect of dispersal limitation on both AM fungal communities and plant phylogenetic lineages, driving high positive spatial auto-correlation. AM fungi are obligate symbionts and, therefore, successful colonization is contingent on the coordinated arrival of fungal spores and suitable plant hosts to new locations. Here it has been shown that the stochastic dispersal processes seem to be important in the assembly of plant-AM fungal communities, but these dispersal processes may be correlated with traits in both AM fungi and their host plants that are influenced by phylogenetic history.
-
A recent study revealed the dominant role of stochasticity in structuring AM fungal communities on a regional scale, with drift generally being the major process[54], which aligned with the present findings. Thus, these results together with previous studies[12,13,55] acknowledge that the relationship between plant phylogeny and AM fungal communities may be scale-dependent, with stronger correlations at local scales, and a strong spatial structuring on regional scales[55]. In common with their plant hosts, AM fungi are essentially sessile and require external vectors. There is evidence for multiple potential dispersal vectors for AM fungi – both abiotic (water and air/wind) and biotic (animals, including humans)[56], e.g., birds are endozoochorous co-dispersers, transporting viable propagules of both partners – plants and AM fungi[56]. As the present study site is a steppe grassland, it is reasonable to speculate that besides wind and wild animals, grazing activities for storing cattle and sheep could be important in generating the regional distribution pattern of AM fungi. This was supported by that grazing substantially increased the stochasticity of AM fungal community assembly in the steppe by reducing the deterministic effects of plant richness on a regional scale[57] in favor of those AM fungi with disturbance-tolerant traits.
Due to the vast area of the Xilingol grassland, the 10 sampling sites in this study might not fully represent the AM fungal diversity of the whole grassland. Further, there is a lack of comparative studies with AM fungal communities in other grassland areas or different ecosystems (e.g. forest) which might limit the understanding of the driving mechanism of AM fungal community assemblage. Third, as sampling was manipulated at only one time, it may be limited to specific seasons and do not reflect the dynamics of arbuscular mycorrhizal fungal communities at different time scales. Thus, for future studies, sampling should be manipulated with more sites in different ecosystems and various temporal dynamics to detect the phylogenetic signal of plant-AM fungal interactions, and to test whether the dominant role of stochasticity in structuring AM fungal communities on the regional scale is a generalized rule or not.
-
The present results showed that geographic distance was the most important factor explaining the phylogenetic β-diversity in AM fungal communities, while the effects of plant phylogeny and environmental variables was not significant. This suggests that at the landscape scale, AMF community structure is largely determined by stochastic events (e.g., dispersal limitation). Furthermore, significant correlations were found between plant phylogeny and the β-MNTD of AMF communities, but this correlation was attenuated after controlling for geographic distance and environmental variables. This suggests that the role of dispersal limitation in influencing the relationship between AM fungi and host plants may be related to the phylogenetic history of plants and AM fungi. This study provides new insights into understanding the evolution and ecology of the plant-AM fungal symbiosis and highlights the important role of dispersal limitation in AM fungal community structure.
-
The authors confirm contribution to the paper as follows: study conception and design: Zhang Q, Zhou J, Chen X; data collection: Zhang Q, Duan J, Xing J, Du S, Yu M, Qi G; analysis and interpretation of results: Zhang Q, Duan J, Goberna M, Jin Y, Chu J, Yang H, Chen X; draft manuscript preparation: Zhang Q, Duan J, Yang H. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
This work was supported by the Central Public-interest Scientific Institution Basal Research Fund (CAFYBB2019QB001, CAFYBB2020ZB001), the Natural Science Foundation of China (Grant No. 31870099) and the Strategic Priority Research Program of Chinese Academy of Sciences (XDA26020102).
-
The authors declare that they have no conflict of interest.
- Supplementary Table S1 Brief description of the sampling sites in a steppe grassland in northern China.
- Supplementary Fig. S1 Plant phylogenetic tree with the tree in Zanne et al. (2014) as the backbone. Numbers are estimated divergence time (million years).
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of Yunnan Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang Q, Duan J, Goberna M, Jin Y, Xing J, et al. 2024. Phylogenetic turnover of arbuscular mycorrhizal fungal communities across steppe grasslands. Agrobiodiversity 1(2): 16−22 doi: 10.48130/abd-0024-0004 

Phylogenetic turnover of arbuscular mycorrhizal fungal communities across steppe grasslands
- Received: 06 July 2024
- Revised: 22 September 2024
- Accepted: 14 October 2024
- Published online: 30 October 2024
Abstract: Ecological interactions are evolutionarily conserved, indicating a tendency of closely related species to interact with similar partners. Arbuscular mycorrhizal (AM) fungi form obligate symbioses with the roots of most land plants. Local host preference is frequently reported as a factor in structuring AM fungal communities. There lacks study about whether AM fungi-host preference could structure AM fungal communities at the regional scales. Here, AM fungal communities of 296 root samples were revealed, encompassing 76 plant species from 29 plant families, which were sampled in steppe in the Xilingol Grassland in northern China. The relative importance of plant phylogeny, geographical distance, and environmental variables were characterized on phylogenetic turnover of AM fungal communities with GLMM-MCMC (the generalized linear mixed model using Markov chain Monte Carlo) and Mantel test approaches. Geographic distance appeared to be more important to the phylogenetic turnover of AM fungal communities than plant phylogeny and environmental variables, evidencing the role of dispersal limitation in shaping the root AM fungal communities. A great majority of phylogenetic beta diversity (betaNTI and betaNRI) is distributed between −2 and +2, which also suggested a random pattern of AM fungal communities. Here, empirical evidence supporting that dispersal limitation is the main determinant of AM fungal communities at the landscape scale is provided and it is suggested that AM fungal communities are mainly structured by stochastic events.