









-
Asparagus cochinchinensis is a well-known medicinal plant within the genus Asparagus in the family Liliaceae. Species in the Asparagus genus are extensively utilized in both pharmacology and food industries[1,2]. Recently, A. cochinchinensis has been recognized as an edible and medicinal herb in China, resulting in an upward trend in market demand. According to the recommendation of the Chinese Pharmacopoeia (2020), Asparagi Radix is derived from the dried root and tuber of Asparagus cochinchinensis (Lour.) Merr.[3]. Modern pharmacology studies have reported that A. cochinchinensis shows cough-suppressing and expectorant properties, which aids in managing airway inflammation and other related conditions[4]. However, previous studies mainly focused on its botanical, traditional, phytochemical, and pharmacological aspects[5,6]. Currently, A. cochinchinensis is mainly planted in Sichuan Province (Neijiang City) and Guangxi Zhuang Autonomous Region. There are few studies discussing the topic regarding its sustainable cultivation practices. Cultivation as the initial stage of the whole medicinal plant production chain, remarkably influences its quality and warrants further investigation. In recent years, it has been reported that the rhizosphere microbiome greatly contributes to the growth and development of medicinal plants[7]. Qu et al. studied the root secretion metabolites and soil microbiome of five medicinal plants. They found that the bacterial and fungal profiles of these five medicinal plant rhizosphere soil samples were biologically distinct[8]. Among these metabolites, ten root secretion metabolites significantly influenced the distribution of bacteria and fungi[9]. Liu et al. studied the rhizosphere microbiome of Glehnia littoralis and observed that the rhizosphere microbial community formed a complex symbiotic network that positively impacted the growth and development of G. littoralis[10]. Wang et al. explored the effect of cultivation process on Atractylodes lancea, and they indicated that the autotoxic allelopathic substances in the rhizosphere of A. lancea altered rhizosphere soil microbiome, leading to continuous cropping disorders[11]. These studies collectively suggest that the soil bacterial community significantly affect the growth and development of medicinal plants. Additionally, the rhizosphere microbiome might also influence the natural product synthesis of medicinal plants. Zhu et al. analyzed the relationship between the rhizosphere bacterial community and the accumulation of polysaccharides of Dendrobium officinale, and they indicated that the relative abundance of Pandoraea, which was identified from the rhizosphere bacterial community, was associated with polysaccharide level. Pandoraea might promote the production of D. officinale polysaccharide production[12]. The function of rhizosphere species was also reported. Ng et al. studied the promotive effect of Bacillus subtilis and Pseudomonas fluorescens on the polysaccharide level of Pseudostellaria heterophylla. Results showed that the polysaccharide level increased by 38% and 253% after incubation with these strains[13]. Based on the above studies, rhizosphere microbiome not only affect the growth and development of medicinal plant, but might also be related to the synthesis of active compounds. Therefore, it is crucial to reveal the rhizosphere microbiome structure and discuss the potential relationship between the rhizosphere microbiome and A. cochinchinensis.
In this study, we simultaneously applied amplicon sequencing and transcriptomic sequencing technology to analyze the rhizosphere microbiome structure and gene expression in A. cochinchinensis. This work advances our understanding of the rhizosphere microbiome resource and provides references for the development of functional rhizosphere strains for A. cochinchinensis.
-
The samples of A. cochinchinensis and their associated rhizosphere soil were collected from Neijiang City, Sichuan Province, China, with detailed collection information provided in Table 1. It has been reported that Neijiang City has an average annual temperature ranging from 15−28 °C and the annual rainfall is about 1,000 mm. The plant samples were used for transcriptomic analysis. After collection, the tender stems and tuberous roots were excised, and the surface of the samples were washed with RNase-free water. The samples were then immediately dried using absorbent paper and cut into 50−100 mg pieces. These pieces were rapidly frozen in liquid nitrogen and placed into pre-cooled threaded Eppendorf tubes, where they were stored at −80 °C. All sampling tools were sterilized and disinfected before using. The rhizosphere soil samples were collected from three towns including Yangjia Town (YRB), Shuangcai Town (CRB), and Guobei Town (SRB), Neijiang City. The soil was removed from the roots, followed by the collection of soil tightly adhering to the roots using a sterile brush. The rhizosphere soil (within 0.2 cm of the roots) was filtered through a 2 mm sieve, and samples from each area were mixed. Next, plant debris was removed and carefully placed into sterilized bags. All soil samples were rapidly frozen in liquid nitrogen and stored at −80 °C until further analysis.
Table 1. Information for the A. cochinchinensis cultivation soil samples and different tissues used in this study.
Sample number Group Sampling date Sampling area SAMN number YRBR1 YRBR 2024/4/24 China: Neiiang: Yangjia Town 29.525142N 105.163067E SAMN43440143 YRBR2 YRBR 2024/4/24 China: Neiiang: Yangjia Town 29.525142N 105.163067E SAMN43440144 YRBR3 YRBR 2024/4/24 China: Neiiang: Yangjia Town 29.525142N 105.163067E SAMN43440145 YRBS1 YRBS 2024/4/24 China: Neiiang: Yangjia Town 29.525142N 105.163067E SAMN43440146 YRBS2 YRBS 2024/4/24 China: Neiiang: Yangjia Town 29.525142N 105.163067E SAMN43440147 YRBS3 YRBS 2024/4/24 China: Neiiang: Yangjia Town29.525142N 105.163067E SAMN43440148 CRB1 CRB 2024/4/24 China: Neiiang: Shuangcai Town 29.749611N 105.109195E SAMN43415523 CRB2 CRB 2024/4/24 China: Neiiang: Shuangcai Town 29.749611N 105.109195E SAMN43415524 CRB3 CRB 2024/4/24 China: Neiiang: Shuangcai Town 29.749611N 105.109195E SAMN43415525 YRB1 YRB 2024/4/24 China: Nelliang: Yangjia Town 29.766105N 105.353238E SAMN43415526 YRB2 YRB 2024/4/24 China: Nelliang: Yangjia Town 29.766105N 105.353238E SAMN43415527 YRB3 YRB 2024/4/24 China: Nelliang: Yangjia Town 29.766105N 105.353238E SAMN43415528 SRB1 SRB 2024/4/24 China: Neiiang: Guobei Town 29.525142N 105.163067E SAMN43415529 SRB2 SRB 2024/4/24 China: Neiiang: Guobei Town 29.525142N 105.163067E SAMN43415530 SRB3 SRB 2024/4/24 China: Neiiang: Guobei Town 29.525142N 105.163067E SAMN43415531 High throughput sequencing of the rhizosphere bacterial community
-
Approximately 1.0 g of frozen soil sample was transferred into a sterilized 15 ml centrifuge tube containing 1.5 g of grinding beads. Bacterial DNA extraction was conducted using the EZNA® Soil DNA Kit (D5625, Omega Bio-Tek, Inc.) according to the manufacturer's protocol. The extracted DNA was quantified using a Nanodrop spectrophotometer (ND ONE, Genes Ltd.). Primers were designed based on conserved regions within the 16S rRNA gene and incorporating sample-specific barcode sequences. The bacterial 16S rRNA gene was amplified by PCR using the primers 16S V3V4-F 5'-CCTACGGGGNGGCWGCAG-3' and 16S V3V4-R 5'-GACTACHVGGGGTATCTAATCC-3'. The PCR products were purified using magnetic bead-based purification methods. Fluorescence-based quantification was performed on the recovered PCR products. Based on the quantification results, samples were mixed in appropriate proportions based on the sequencing requirements. Library preparation was conducted using the VAHTS Universal DNA Library Prep Kit for Illumina V3 and VAHTS DNA Adapters set3-set6 for Illumina from Novogene. The libraries were subjected to quality control and sequenced on the NovaSeq 6000 platform. Barcodes were used to identify the mixed samples and obtain individual sample sequence data. Low quality reads were filtered using Trimmomatic software (Version 0.39), with a window size of 20 bp and an average quality value greater than 20 within the window. Primer sequences were identified and removed using cutadapt software (Version 3.5), resulting in clean sequences. Each sequence generated by DADA2 was termed a feature or amplicon sequence variant (ASV). Low abundance ASVs were filtered using QIIME2 (Version 2022.3), and each ASV was annotated at various taxonomies. This process enabled the determination of bacterial community composition within the samples. R software (Version 3.1.1) with relevant packages was used to evaluate the alpha diversity and beta diversity based on the Bray-Curtis distance. The bacterial composition at the phylum, class, order, family, and genus levels were displayed using the barplot package, which was conducted to reveal differences between samples. Additionally, based on the Spearman correlation matrix, co-occurrence network topology analysis was conducted using the R Igraph package.
RNA extraction, cDNA library construction, sequencing, de novo assembly, and annotation
-
Total RNA was extracted from the tuberous roots and stems of A. cochinchinensis for cDNA library construction, with each treatment performed in triplicate. RNA integrity and quality were evaluated using the NanoDrop One spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and a Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). mRNA was enriched using oligo (dT) magnetic beads, and double-stranded cDNA was synthesized. The library preparation was completed using the MGIEasy RNA Library Prep Kit for BGI. The fragment size and concentration of the library were assessed using an Agilent 2100 Bioanalyzer. Sequencing was performed on the BGI high-throughput sequencing platform DNBSEQ-T7. After filtering out raw data connections, poly-N sequences, and low-quality reads, clean reads were obtained. To obtain valid data for subsequent analyses, the raw data was filtered with fastp (Version 0.21.0) and fastqc (Version 0.11.9)[14,15]. De novo assembly was conducted using Trinity (Version 2.11.0), and resulting transcripts were clustered using cd-hit (Version 4.8.1) to obtain universal genes (unigenes)[16,17]. The unigenes were annotated using two methods: 1) sequence similarity search using diamond blastp (Version 2.0.6.144; parameters: e-value 1e-5) for comparison; 2) motif similarity search, structural domain prediction using hmmscan (Version 3.3.2; parameters: e-value 0.01) to obtain conserved sequences, motifs, and structural domains of proteins[18,19]. Comprehensive functional annotation of the unigenes were performed using seven databases: Nr, Pfam, Uniprot, KEGG, GO, KOG/COG, and PATHWAY[20−24].
Differential expression unigene analysis
-
Differential expression analysis was conducted on the counts of expressed unigenes obtained from quantitative expression measurements across various samples. DESeq2 software was used for differential expressed gene analysis (Version 1.26.0)[25]. The significance threshold was initially set to padj < 0.05 and |log2FoldChange| > 1. The padj value was used for analyzing significant differences. The objective was to determine whether the functions of the differentially expressed unigenes were concentrated in specific functional categories. Common functional classifications included GO and KEGG Pathway. The enrichment analysis was performed for hypergeometric tests to identify GO terms and KEGG pathways that were significantly enriched among the differentially expressed unigenes relative to all annotated unigenes. The clusterProfiler software (Version 3.14.3) was utilized for differential expression unigene enrichment analysis[26].
Quantitative real-time -PCR
-
The RNA products of the tuberous roots and stems of A. cochinchinensis were extracted using the FastPure Plant Total RNA Isolation Kit (Polysaccharides & Polyphenolics–rich, RC401-01, Vazyme, Nanjing, China). Reverse transcription was performed using the RevertAid™ First Strand cDNA Synthesis Kit, with DNase I (RT-01022, Thermo Scientific, Waltham, USA). The TB Green® Premix Ex Taq™ II (Tli RNaseH Plus, RR82LR, TaKaRa, Beijing, China) was used to perform qRT-PCR. The mRNA expression of Unigene11685, Unigene10718, Unigene11242, and Unigene10145 were evaluated by the 2−ΔΔCᴛ method and normalized with EF1α. The primer sequences were designed in Sangon Biotech (Sangon, Shanghai, China, Table 2).
Table 2. Designed primer information in this study.
Primers DNA sequence (5' to 3') U10703-F ACCTACCGCCATCACCTCAAC U10703-R CCCGAACCAAATCCCTGAAATACC U11242-F CTTGATATAGACGATCCTGACACTTGG U11242-R TGGGAAGCGATTATTGAAACCTCTG U11685-F ATGGCGGTCTCGGTCATACTAC U11685-R GGCATTGGATTTGGATTTGGATCTG U10145-F GCCAAGTGAGTTGCCAGGTTC U10145-R GTTTATGTATCAGGTCGTCTTGCTTTG U10718 -F GGTAACTTGGCACTTAAAGCGATATAG U10718 -R TACCTTCTCGTGTACCATTAACAACTC EF1α-F CTGGCCAGGGTGGTTCATGAT EF1α-R TAAGTCTGTTGAGATGCACC Statistical analysis
-
The data in this study were subjected to statistical testing with SPSS software (Version 22.0) and RStudio (Version 2024.04.2+764). Significant differences were analyzed by one-way ANOVA. A p-value less than 0.05 (p < 0.05) in these analyses indicated a significant difference.
-
In this study, high-throughput sequencing technology was employed to analyze the rhizosphere bacterial composition of samples collected from three areas. A total of 3,372,071 raw reads were observed from the nine samples. After quality control, 3,181,644 clean reads were obtained. The raw sequencing data was uploaded to the National Center for Biotechnology Information Sequence Read Archive database with accession numbers CRB1 (SAMN43415523), CRB2 (SAMN43415524), CRB3 (SAMN43415525), YRB1 (SAMN43415526), YRB2 (SAMN43415527), YRB3 (SAMN43415528), SRB1 (SAMN43415529), SRB2 (SAMN43415530), and SRB3 (SAMN43415531). Rarefaction curve analysis results indicated that the sequencing depth in each sample was sufficient to reflect the bacterial community (Fig. 1a). A venn diagram shows the number of shared or unique ASVs across different groups, revealing 2579 shared ASVs in various groups. The numbers of unique ASVs in the CRB, SRB, and YRB groups were 8654, 13093, and 9093, respectively. Notably, the SRB group exhibited the highest number of unique ASVs, while the CRB group had the lowest number (Fig. 1b). The alpha diversity analysis was performed to evaluate the richness and diversity of the bacterial communities. The indices of alpha diversity are listed in Supplementary Table S1. The average ACE indices for CRB, SRB, and YRB groups were 6,982, 8,181, and 7,615, respectively (Fig. 1c). The average Chao1 indices were 6,960, 8,156, and 7,599 (Fig. 1d). The average Shannon indices were 11.08, 11.67, and 11.51, while the average Simpson indices were 0.996, 0.999, and 0.999 (Fig. 1e, f). No significant differences were observed in the Shannon, Simpson, Chao1, and ACE indices among various groups. The SRB group exhibited the highest Shannon, Chao1, and ACE indices followed by the YRB group, while the CRB group consistently showed the lowest values across these indices.
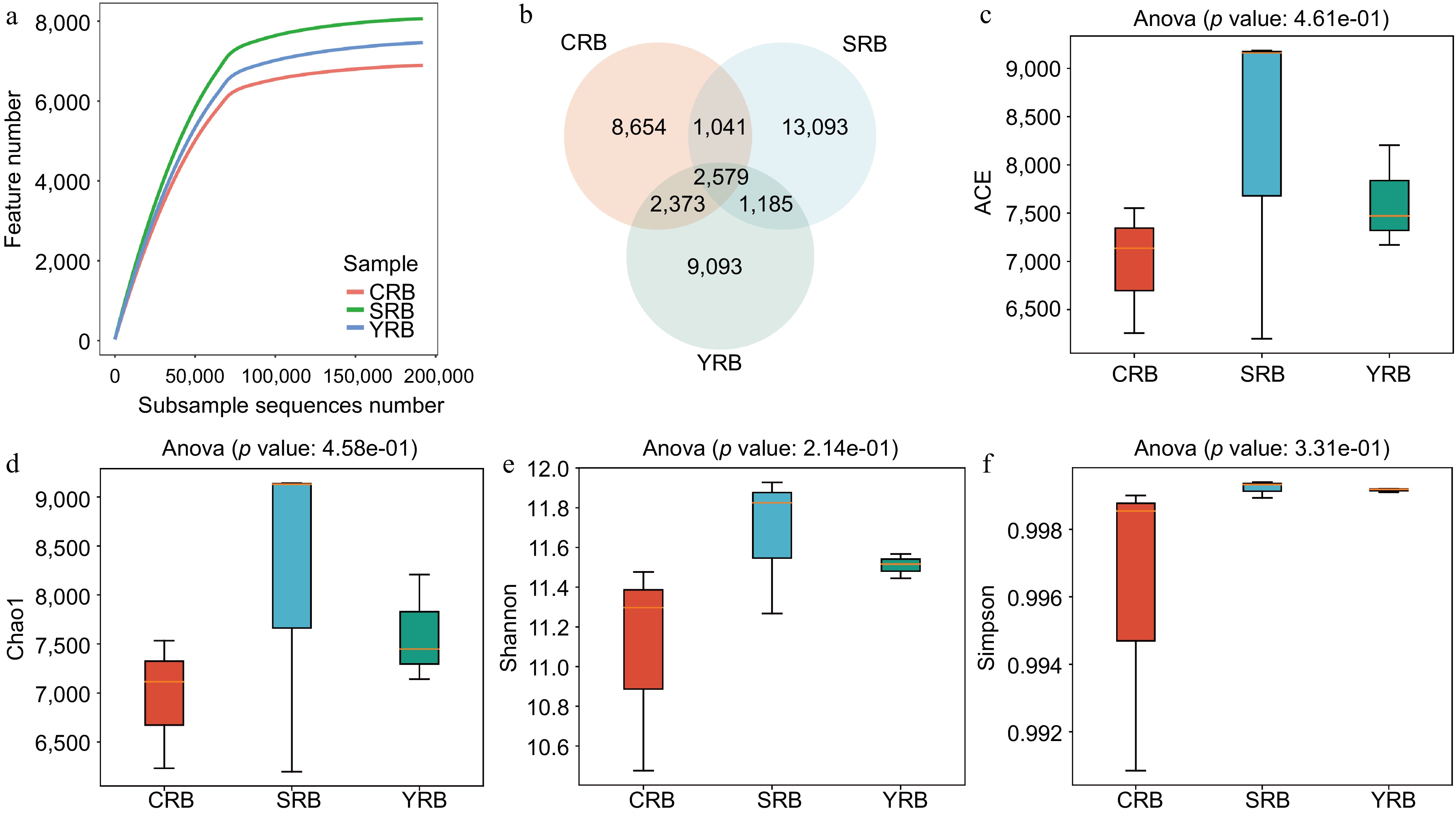
Figure 1.
The diversity and richness of rhizosphere bacterial communities of A. cochinchinensis rhizosphere samples collected from various areas. (a) Rarefaction curve reveals the sequencing depth in each sample, the horizontal coordinate reflects the number of sequences randomly selected, and the vertical coordinate reflects the number of features obtained based on the number of sequences. Each curve represents a sample/group and is marked with a different color. (b) Venn diagram illustrates the numbers of ASVs in the CRB, SRB, and YRB groups. (c) ACE indices in the CRB, SRB, and YRB groups based on QIIME2 software, the horizontal coordinate reflects the group name, and the vertical coordinate reflects the ACE index value. (d) Chao1 indices in the CRB, SRB, and YRB groups based on QIIME2 software, the horizontal coordinate reflects the group name, and the vertical coordinate reflects the Chao1 index value.(e) Shannon indices in the CRB, SRB, and YRB groups based on QIIME2 software, the horizontal coordinate reflects the group name, and the vertical coordinate reflects the Shannon index value. (f) Simpson indices in the CRB, SRB, and YRB groups based on QIIME2 software, the horizontal coordinate reflects the group name, and the vertical coordinate reflects the Simpson index value.
Bacterial community composition in A. cochinchinensis rhizosphere soil samples collected from various areas
-
Based on the high-throughput sequencing data, a total of 50 phyla, 138 classes, 343 orders, 490 families, and 875 genera were identified in this study. At the phylum level, the dominant phyla included Proteobacteria, Actinobacteria, Planctomycetota, Actinobacteriota, Chloroflexi, Gemmatimonadota, Bacteroidota, Verrucomicrobiota, Myxococcota, and Methylomirabilota with relative abundances greater than 1% (Fig. 2a). Among these phyla, Proteobacteria, Actinobacteria, and Planctomycetota were dominant, with relative abundances ranging from 17.8%−23.1%, 14.8%−24.4%, and 8.2%−12.6%, respectively. The most abundant bacterial classes in this study included Gammaproteobacteria, Vicinamibacteria, Alphaproteobacteria, Phycisphaerae, Blastocatellia, Bacteroidia, Planctomycetes, Gemmatimonadetes, Thermoleophilia, and Verrucomicrobiae. Gammaproteobacteria, Vicinamibacteria, and Alphaproteobacteria were dominant, with relative abundances of 10.2%−12.8%, 6.7%−17.0%, and 6.3%−12.6%, respectively (Fig. 2b). At the order level, Vicinamibacterales had the highest relative abundance, while the CRB group had lower levels of Vicinamibacterales compared to the other groups, the SRB group had a lower relative abundance of Pyrinomonadales compared to the other groups (Fig. 2c). At the family level, Gemmatimonadaceae had the highest relative abundance, with the highest relative abundance in the CRB group, whereas Nitrosomonadaceae had the highest average relative abundance in the YRB group. Other abundant families included Gemmatimonadaceae, Nitrosomonadaceae, Vicinamibacteraceae, Pyrinomonadaceae, WD2101_soil_group, and Sphingomonadaceaes (Fig. 2d). At the genus level, RB41 had the highest relative abundance, with the highest average relative abundance in the CRB group. RB41, Vicinamibacteraceae, and WD2101_soil had relative abundances ranging from 1.7%−6.7%, 2.6%−4.8%, and 1.8%−6.0%, respectively (Fig. 2e).
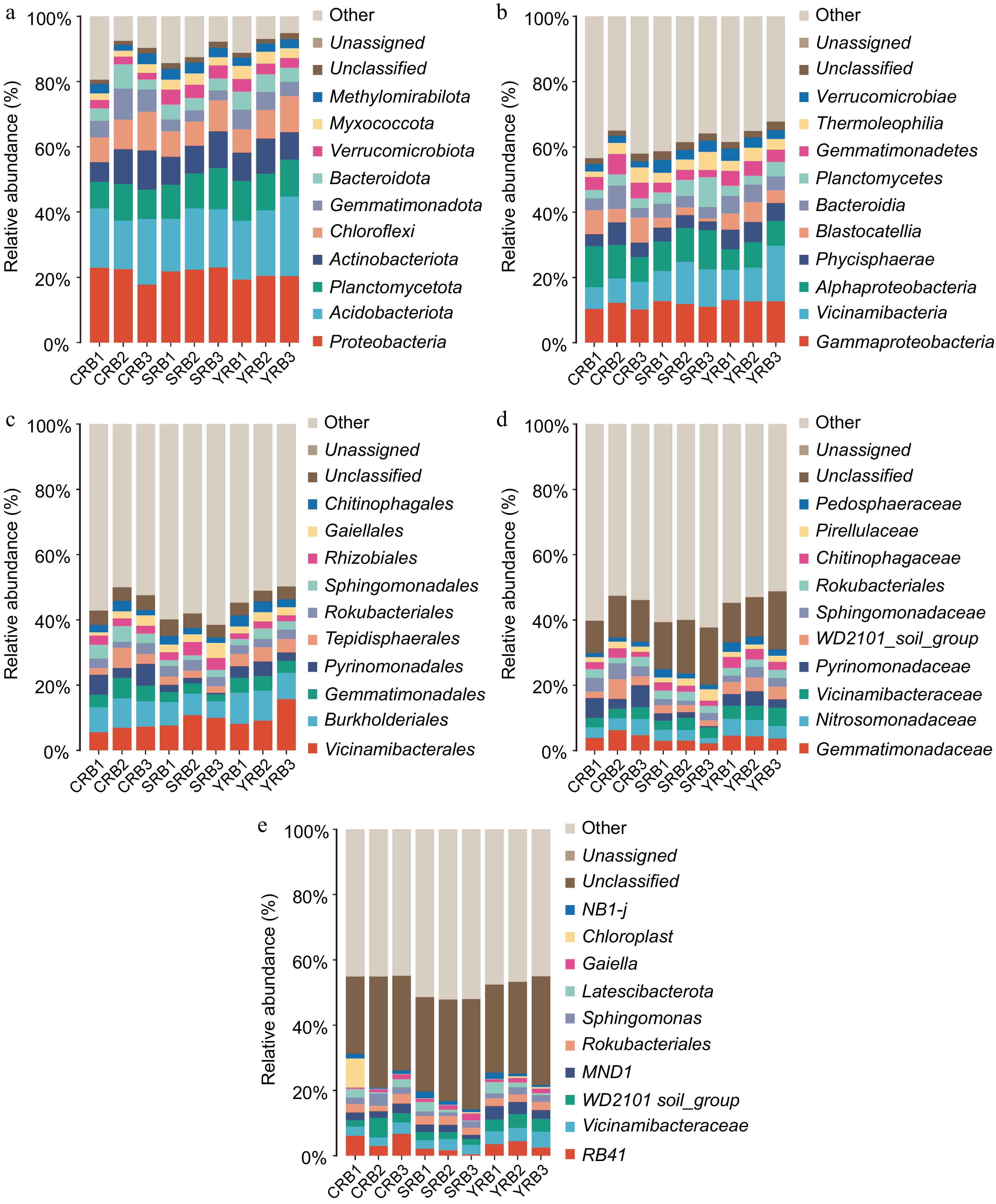
Figure 2.
Bacterial community composition in the A. cochinchinensis rhizosphere samples collected from various areas. (a) The relative abundances of the ten top dominant taxa at the phylum level. (b) The relative abundances of the ten top dominant taxa at the class level. (c) The relative abundances of the ten top dominant taxa at the order level. (d) The relative abundances of the ten top dominant taxa at the family level. (e) The relative abundances of the ten top dominant taxa at the genus level.
Differences of bacterial community of A. cochinchinensis rhizosphere soil samples collected from various areas
-
PCA, PCoA, and NMDS analyses were used to assess the similarity and differences between various groups. Using the binary Jaccard algorithm, the first component in the PCA plot explained 23.25% of the variance, with the CRB, SRB, and YRB groups clustering separately (Fig. 3a). Similarly, based on the PCoA analysis result, the first component explained 21.78% of the variance, and the three groups also formed distinct clusters (Fig. 3b). Based on the NMDS analysis result, CRB, SRB, and YRB groups were clustered separately, and the bacterial community composition was similar between the YRB and CRB groups (Fig. 3c). According to the Bray-Curtis distance algorithm and average clustering for bacterial community analysis at the genus level, the hierarchical clustering tree further revealed that the YRB and CRB groups were clustered, while SRB group formed a separate cluster (Fig. 3d). The histogram indicated that the bacterial communities at the genus level were different, the dominant bacterial composition was similar with differences in relative abundance. The heatmap also further supported this result (Fig. 3e).
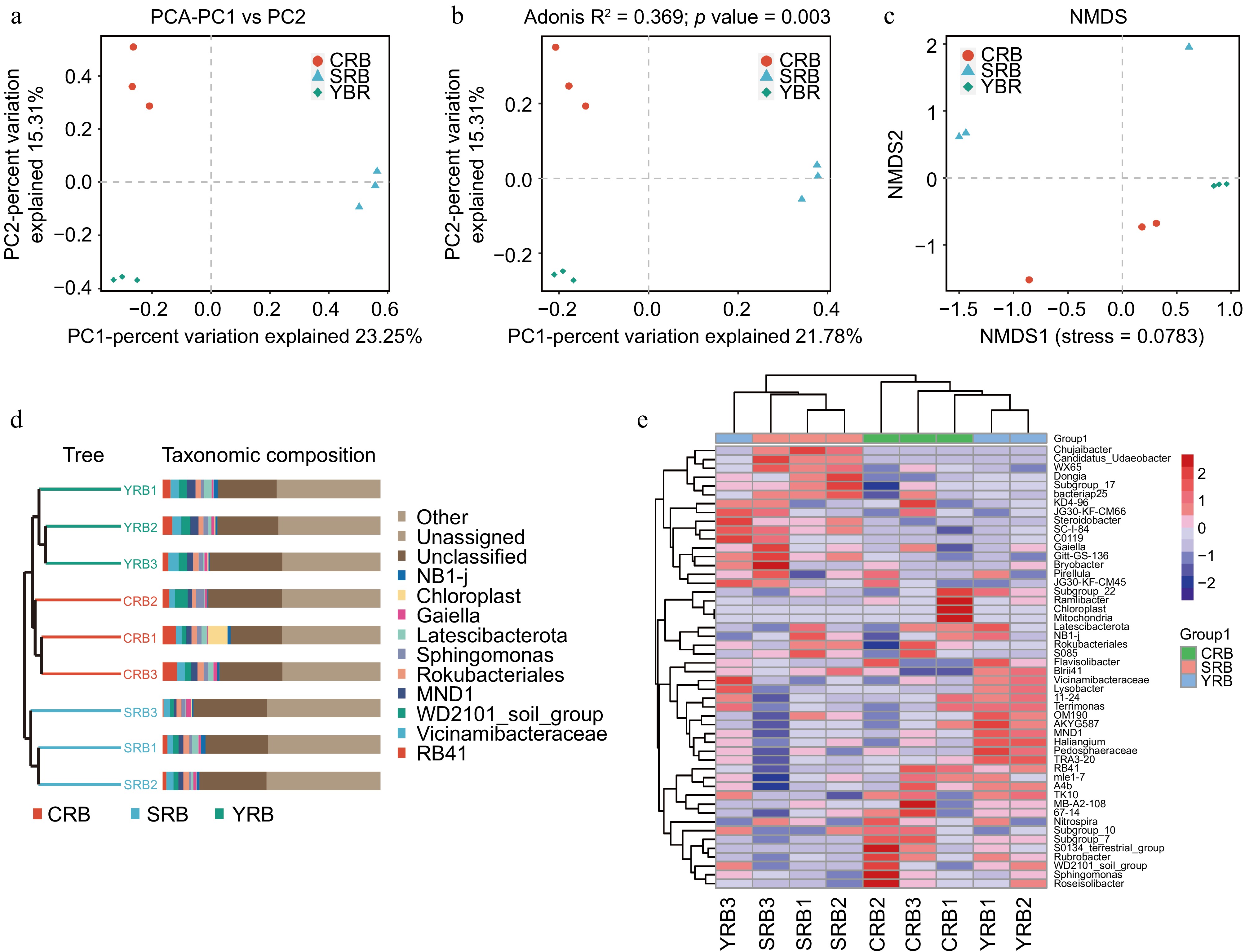
Figure 3.
Beta diversity of bacterial community in the A. cochinchinensis rhizosphere samples collected from various areas. (a) Principal component analysis of gut bacterial community among different groups. (b) Principal co-ordinates analysis of soil bacterial community among different groups. (c) Non-metric multi-dimensional scaling analysis of soil bacterial community among different groups. (d) Unweighted pair-group method with arithmetic mean combined with species abundance histograms analysis among different groups based on the binary Jaccard distances. (e) Heatmap clustering at the genus level and the top 50 species among different groups.
Correlation of the bacterial community of A. cochinchinensis rhizosphere soil samples collected from various areas
-
To explore the interactions between bacterial taxa in different groups, correlation network graphs were constructed to analyze the interaction between the top 50 abundant taxa at the genus level across all samples. These networks consisted of an average of 50 nodes and 472 connecting lines. From the results of the analysis, RB41 and Niastella in the CRB group were positively correlated to the whole bacterial community, while Subgroup_7 and Rubrobacter were negatively correlated to the whole bacterial community (Fig. 4a). In the YRB group, Rubrobacter, and Gaiella were positively correlated with the whole bacterial community, while TK1O showed negative correlation, and Candidatus_Udaeobacter and Gaiella were positively correlated with the whole bacterial community in the SRB group (Fig. 4b, c). JG30-KF-AS9 and Candidatus_Kaiserbacteria showed negative correlation.
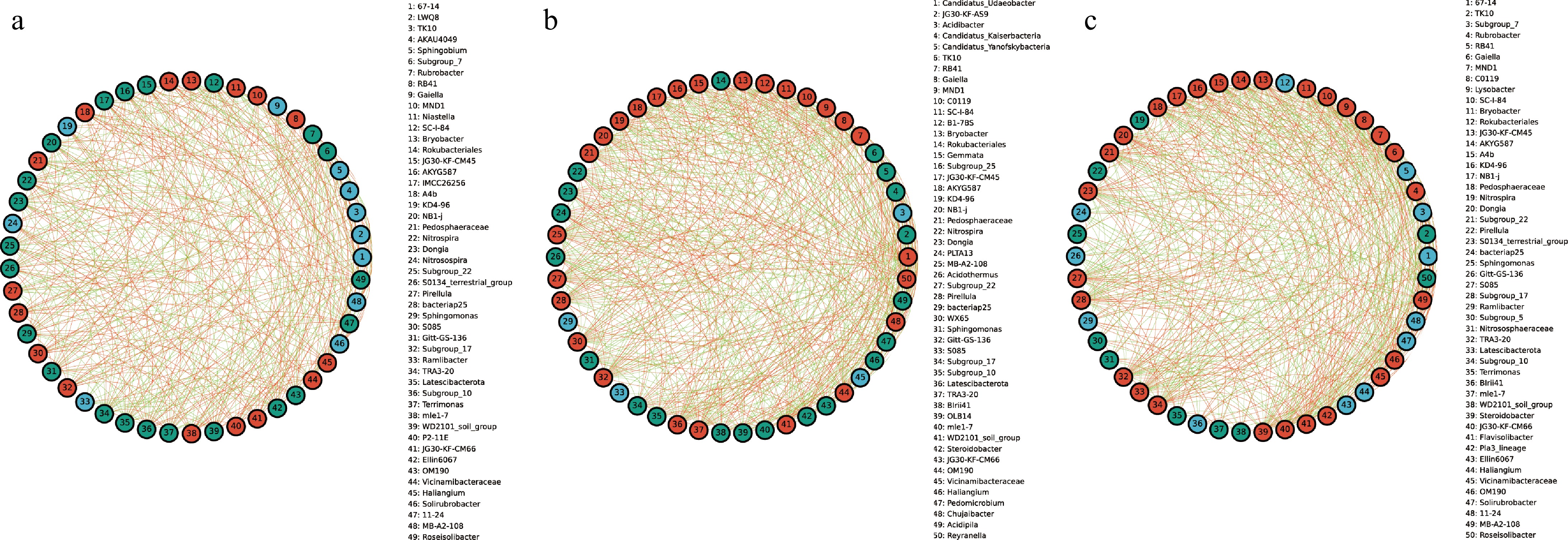
Figure 4.
The species correlation network of bacterial communities of A. cochinchinensis rhizosphere samples collected from various areas. Spearman rank correlation analyses were performed on the top 50 species in the (a) CRB, (b) SRB, and (c) YRB in terms of genus-level abundance, and correlation networks were constructed by filtering data with correlations greater than 0.6 or less than -0.6 and p-values of less than 0.05. The green lines represent negative correlations, and red lines represent positive correlations. The bolder lines represent the closer correlations between two genera.
RNA-seq data revealed the gene expression pattern of A. cochinchinensis
-
RNA-seq analysis of the tuberous roots and stems of A. cochinchinensis was performed. Based on the sequencing result, an average of 6.27 GB of clean data was obtained from 68.91 GB of raw reads from all samples. The GC content was 46%, and the Q30 value was 97%. A total of 240,208,194 raw reads were generated (Supplementary Table S2). After removing low-quality sequences, 240,208,064 clean reads remained. The clean reads were assembled into 254,407 transcripts. The data was uploaded to the National Center for Biotechnology Information Sequence Read Archive database with accession numbers YRBS3 (SAMN43440148), YRBS2 (SAMN43440147), YRBS1 (SAMN43440146), YRBR3 (SAMN43440145), YRBR2 (SAMN43440144), and YRBR1 (SAMN43440143). These transcripts were further clustered using cd-hit (Version 4.8.1)[13], resulting in 127,262 unigene sequences. The sequence length ranged from 201 to 16,535 bp, with an average length of 910.2 bp and an N50 length of 1,661 bp. The GO database was used to categorize the functions of predicted unigenes into three categories: cellular component (CC), molecular function (MF), and biological process (BP)[17]. Of the 23,299 unigenes corresponding to known protein families, the top 20 annotations in each GO slim secondary classification were selected for analysis. The function of most unigenes were predicted for cellular components, followed by molecular functions and biological processes. Within cellular components, the unigenes were predicted for membrane, nucleolus, and cytoplasm. For biological processes, the unigenes were predicted for phosphorylation, proteolysis, and regulation of DNA-templated transcription. For molecular functions, the unigenes were predicted for ATP binding, metal ion binding, and DNA binding (Fig. 5a). KEGG annotation of unigene sequences categorized the pathways involving the largest number of unique transcripts as global and overview maps, involved in carbohydrate metabolism, amino acid metabolism, and translation, transcription, and signal transduction pathways (Fig. 5b).
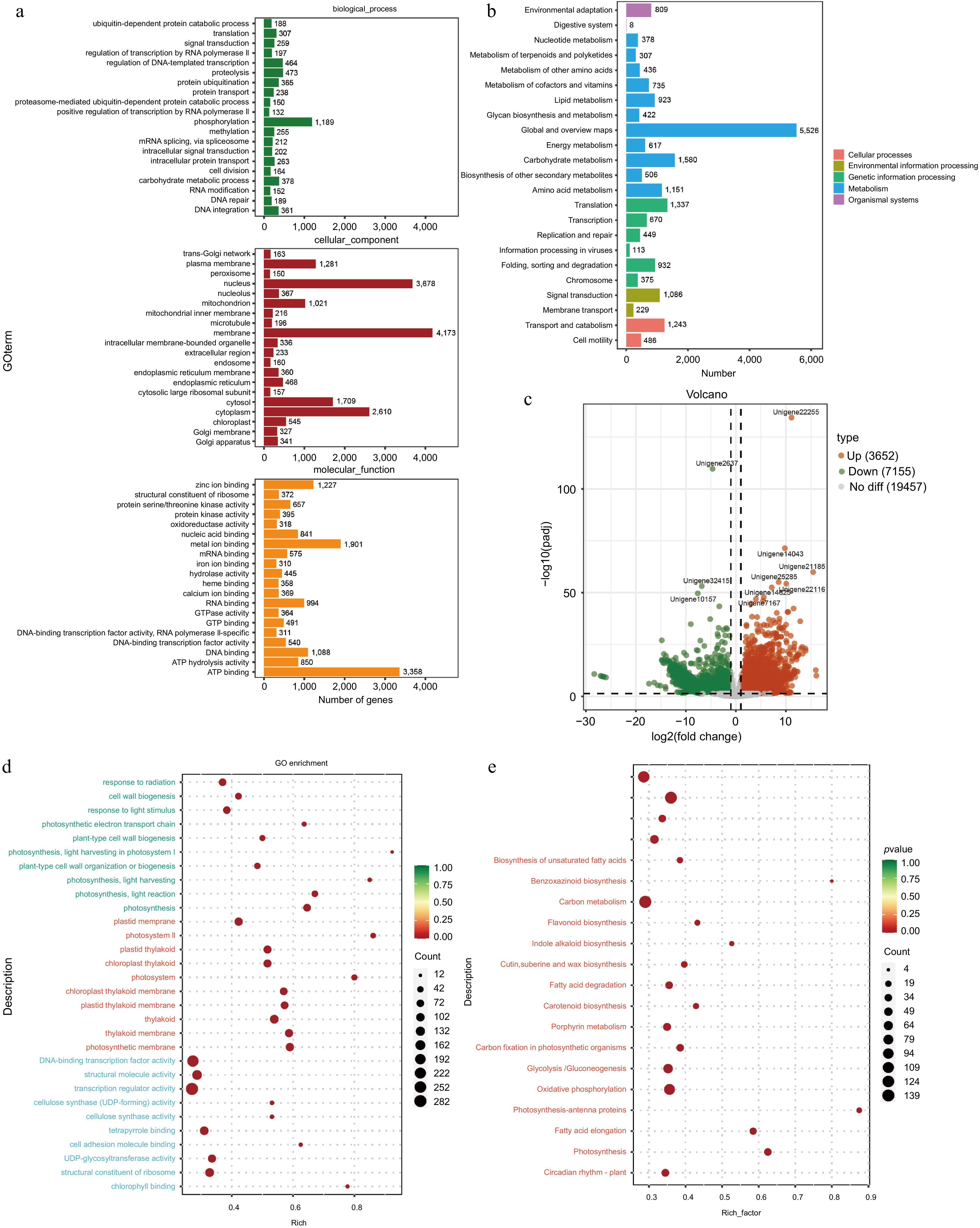
Figure 5.
Functional prediction of different tissues of A. cochinchinensis. (a) Function prediction of different tissues of A. cochinchinensis based on the Gene Ontology database. (b) Function prediction of different tissues of A. cochinchinensis based on the Kyoto Encyclopedia of Genes and Genomes database. (c) Comparing the differential expression of roots and stems unigene. (d) Gene function prediction of differentially expressed unigenes based on the Gene Ontology database. (e) Gene function prediction of differentially expressed unigenes based on the Kyoto Encyclopedia of Genes and Genomes database.
Differentially expressed gene analysis between tuberous roots and stem tissues in A. cochinchinensis
-
For the differential expression analysis of unigenes across various tissues, the threshold of padj < 0.05 and |log2FoldChange| > 1 was applied. The difference in unigene expression between the stem and tuberous root tissues were observed. Specifically, with a total of 10,807 differentially expressed unigenes between the stem and tuberous roots tissues, with 3,652 unigenes upregulated and 7,155 genes downregulated in tuberous roots compared to stems (Fig. 5c). Functional analysis based on the GO and KEGG databases revealed that the most enriched GO pathways in tuberous roots involved DNA-binding transcription factor activity, cellulose synthase (UDP-forming) activity, and cell wall biogenesis (Fig. 5d). KEGG analysis indicated enrichment in pathways including ribosome, pentose phosphate pathway, fatty acid metabolism, and glycolysis /gluconeogenesis (Fig. 5e). Based on the RNA-seq data, four differentially expressed unigenes related to the growth and biotic stress resistance between the tuberous root and stem samples of A. cochinchinensis. Significant differences of all these four genes were observed through qPCR results, which supported transcriptomics data (Fig. 6).
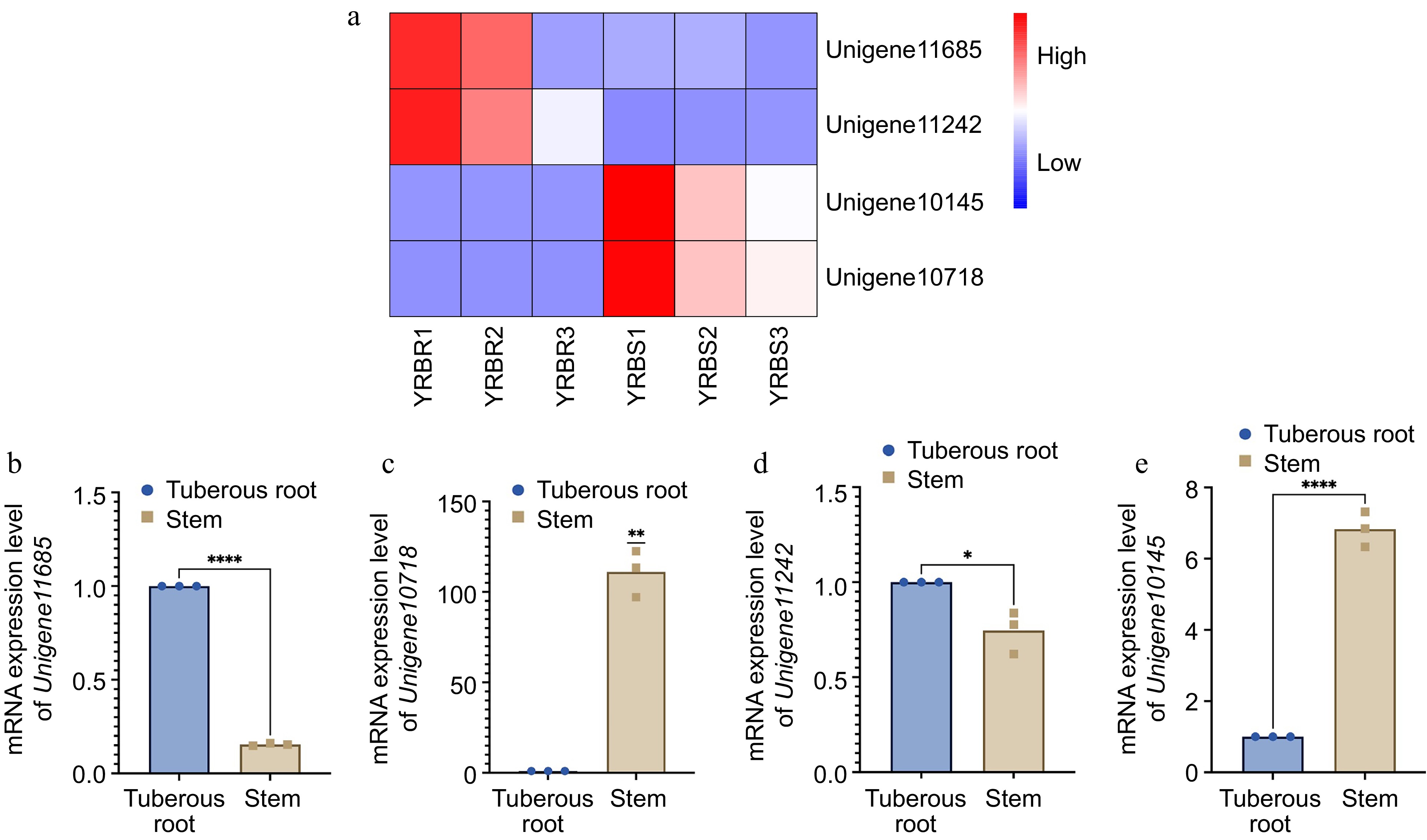
Figure 6.
mRNA expression level of four differentially expressed genes in different tissues of A. cochinchinensis. (a) Comparison of expression level of Unigene11685, Unigene10718, Unigene11242, and Unigene10145 in tuberous root and stem samples of A. cochinchinensis based on heatmap. (b) mRNA expression level of Unigene11685 normalized with EF1α in the tuberous root and stem samples of A. cochinchinensis. (c) mRNA expression level of Unigene10718 normalized with EF1α in the tuberous root and stem samples of A. cochinchinensis. (d) mRNA expression level of Unigene11242 normalized with EF1α in the tuberous root and stem samples of A. cochinchinensis. (e) mRNA expression level of Unigene10145 normalized with EF1α in the tuberous root and stem samples of A. cochinchinensis. * p < 0.05, ** p < 0.01, **** p < 0.0001.
-
The topic of the rhizosphere microbiome has attracted wide attention, which mainly discusses the interaction between plant roots and the rhizosphere microbial community involving bacteria, fungi, archaea, protists, and viruses. Current studies have demonstrated that the composition and characteristics of the rhizosphere microbiome exhibits specificity across different plants. Li et al. studied the rhizosphere microbial community of Astragalus membranaceus and found that the dominant bacterial phyla in the roots of A. membranaceus were Proteobacteria and Actinobacteria[27]. Li et al. analyzed the microbial community of Panax notoginseng, indicating that Chloroflexi and Verrucomicrobia were dominant[28]. Furthermore, Zuo et al. observed differences in the composition of bacterial phyla when comparing the rhizosphere microbial community of Dendrobium plants. These findings underscore the specificity of rhizosphere microbial communities in different plant species[29]. Additionally, the function of rhizosphere microbiome exhibits differences under various conditions. Zhang et al. reported a close relationship between ecological environments and bacterial population structures. Their research on ginseng cultivated in farmland and forests revealed notable differences in the gene functions of the rhizosphere communities between ginseng samples grown in these two environments, which played significant roles in promoting the growth, development, and health of ginseng[30]. Meanwhile, Jiao et al. indicated that heavy metals inhibited the growth of corn, and affected the diversity of corn rhizosphere soil microbial community, as well as their metabolic pathways, and metabolites[31]. Liu et al. conducted precipitation control experiments on four plant species and discovered that microbial functions were primarily influenced by water availability rather than being species-dependent. Changes in water levels caused by precipitation may impact the element cycle of ecosystems by altering the soil microbial community[32]. Furthermore, De Francisco Martínez et al. identified novel cold-tolerance-related genes from the rhizosphere microorganisms of two Antarctic cold-tolerant plants[33]. These findings collectively demonstrate the influence of rhizosphere microorganisms on different host plants across various habitats and the functional diversity of microbial communities. Thus, it is essential to reveal the relationship between the rhizosphere microbiome and host plants to provide a basis for optimizing the cultivation process in agricultural production systems. In this study, we employed amplicon sequencing and transcriptomics technology to investigate the rhizosphere bacterial community structure of A. cochinchinensis and analyze the gene expression of host plants. Results showed that RB41, Vicinamibacteraceae, and WD2101_soil, and all these bacterial genera were common in medicinal plants, which was similar with previous studies[34−38]. Additionally, the transcriptomics data demonstrated that the rhizosphere bacterial community not only promoted the growth of A. cochinchinensis but also assisted the host plants in resisting the biotic and abiotic stress. It has been reported that the rhizosphere microbiome has the potential to influence natural product synthesis in medicinal plants. The rhizosphere is an important place for compound exchange between medicinal plants and soil[39,40]. Rhizosphere microorganisms had an important impact on the growth and the formation of bioactive compounds[41]. Jamwal et al. observed that the metabolites and rhizosphere microbial community in plants of Coleus barbatus in different developmental stages are changing[42]. Similarly, Chen et al. demonstrated that the composition of rhizosphere microorganisms had a profound influence on key bioactive compounds, including dicarboxylates, organic acids, and polysaccharides. It has been reported that polysaccharides are the major bioactive compounds in A. cochinchinensis[43]. Fan et al. have highlighted the role of the rhizosphere microbiome in affecting polysaccharide accumulation in Codonopsis pilosula[44]. Ding et al. conducted metagenomic sequencing on the rhizosphere microorganisms of three Astragalus species, revealing that KEGG annotations of these microbial communities were enriched in metabolic pathways related to triterpenoids, flavonoids, and polysaccharides[45]. There are however limited studies focusing on the relationship between A. cochinchinensis polysaccharide synthesis and rhizosphere microbiome. Given these findings, further exploration is warranted to identify beneficial rhizosphere bacteria that may enhance polysaccharide synthesis in A. cochinchinensis. This study offers valuable insights for optimizing the cultivation and medicinal quality of A. cochinchinensis.
This work was supported by introduces the talented person scientific research start funds subsidization project of Chengdu University of Traditional Chinese Medicine (030040015).
-
The authors confirm contribution to the paper as follows: project administration: Zhang X, Song C; resources: Zhang X, Liu X, Tang X, Wang L, Chen J, Liu H, Liang S, Wang X; formal analysis: Zhang X, Yang S, Liu C; methodology: Zhang X, Yu J, Liu X, Tang X, Wang L, Chen J, Luo H, Liang S, Wang X, Song C; data curation: Zhang X; writing – original draft: Zhang X, Yang Y, Yu J, Song C; writing – review & editing: Zhang X, Yu J, Song C; investigation: Yang S; conceptualization, Funding acquisition: Song C. All authors reviewed the results and approved the final version of the manuscript
-
The data that support the findings of this study are available in the National Center for Biotechnology Information Sequence Read Archive database repository with accession numbers SAMN43415523- SAMN43415531, SAMN43440143-SAMN43440148.
-
The authors declare that they have no conflict of interest. Dr. Chi Song is the Editorial Board member of Medicinal Plant Biology who was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal's standard procedures, with peer-review handled independently of this Editorial Board member and the research group.
-
# Authors contributed equally: Xiaoyong Zhang, Shuai Yang, Jingsheng Yu
- Supplementary Table S1 The indices of alpha diversity for soil samples.
- Supplementary Table S2 The amount of sequencing data of different tissues.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang X, Yang S, Yu J, Liu X, Tang X, et al. 2025. Integrated amplicon sequencing and transcriptomic sequencing technology reveals changes in the bacterial community and gene expression in the rhizosphere soil of Asparagus cochinchinensis. Medicinal Plant Biology 4: e003 doi: 10.48130/mpb-0025-0001 

Integrated amplicon sequencing and transcriptomic sequencing technology reveals changes in the bacterial community and gene expression in the rhizosphere soil of Asparagus cochinchinensis
- Received: 27 November 2024
- Revised: 21 December 2024
- Accepted: 01 January 2025
- Published online: 27 February 2025
Abstract: Asparagus cochinchinensis has been recognized as an edible and medicinal herb in China, which has resulted in high market demand. Therefore, optimizing its cultivation practice is essential to maintain and enhance production levels. In this study, we simultaneously employed amplicon sequencing and transcriptomic sequencing technology to analyze the rhizosphere bacterial community structure and gene expression level in A. cochinchinensis collected from the main production areas. Results showed that a total of 50 phyla, 138 classes, 343 orders, 490 families, and 875 genera were identified, of which Proteobacteria and RB41 were the most abundant at the phylum and genus level, respectively. Transcriptomics results showed that the gene expression was related to biotic stress resistance, plant growth and development, and polysaccharide metabolism, which was supported by qPCR results. These findings offer valuable insights into the utilization safety and quality improvement of A. cochinchinensis.