









-
Flower color change during anthesis, or post-anthesis color change (PACC), is a widespread and natural phenomenon, distinct from simple flower degeneration[1,2]. PACC is generally considered an adaptive signal indicating floral suitability to pollinators by redirecting pollinators to a close range while maintaining long-distance attraction to the plant's floral display[3,4]. PACC is also a favorable trait for horticultural plants as the retention of post-change older flowers enhances floral display size and prolongs the ornamental period.
PACC is associated with changes in flower pigment composition, primarily of carotenoids and anthocyanins[5]. Color changes resulting from changes in carotenoid content mainly lead to minor hue changes in yellow and white flowers, such as Lonicera japonica[6]. Conversely, more striking color transitions are often accompanied by shifts in anthocyanin composition. Many plants exhibit PACC because of an increase or decrease in anthocyanin content, resulting in significant color changes; for example, the post-anthesis transition in flower color from white to red in Hisbiscus mutabilis[7], white to pink in Nicotiana mutabilis[8], white to purple in Viola cornuta 'Yesterday, Today and Tomorrow'[9], yellow to red in Lotus filicaulis and Rosa hybrida 'Ehigasa'[1,10], and purple to white in Brunfelsia acuminata[11]. Sunlight and pollination are critical environmental triggers for color changes[9]. For example, petal color change in Quisqualis indica was induced by light[12], and pollination significantly accelerated PACC in Lotus filicaulis and Euphrasia dyeri[1,13].
The biosynthetic pathway and transcriptional regulation of anthocyanins in plants are well documented[14,15], and many structural genes involved in the anthocyanin biosynthesis pathway have been identified in roses[16]. Several transcription factors associated with the regulation of the anthocyanin biosynthetic pathway have also been characterized, and three MYB-bHLH-WD40 (MBW) complexes (RcMYB1-RcbHLH42-RcTTG1, RcMYB1-RcEGL1-RcTTG1, and MYB114a-bHLH3-WD40) have been linked to anthocyanin accumulation[17,18]. Other transcription factors, such as B-box zinc finger (BBX), NAC (NAM, ATAF, and CUC), and WRKY, also affect the transcriptional regulation of anthocyanin biosynthesis in Rosaceae plants[19−21].
Anthocyanins are biosynthesized on the cytoplasmic surface of the endoplasmic reticulum and transported into the vacuole for storage[22]. Many anthocyanin transport genes have been identified, including ATP-binding cassette (ABC), multidrug and toxic compound extrusion (MATE), and glutathione S-transferase (GST) genes[23]. Various GSTs function as anthocyanin transporters in plants, such as ZmBZ2 in maize (Zea mays)[24], PhAN9 in petunia (Petunia hybrida)[25], AtTT19 (AtGSTF12) in Arabidopsis thaliana[26], CkmGST3 in cyclamen (Cyclamen 'Kaori-no-mai')[27], PpRiant (PpGST1) in peach (Prunus persica)[28,29], VvGST4 in grapevine (Vitis vinifera)[30], LcGST4 in litchi (Litchi chinensis)[31], FvRAP in strawberry (Fragaria vesca)[32], CsGSTF1 in purple tea (Camellia sinensis)[33], IbGSTF4 in Ipomoea batatas[34], MdGSTF6 in apple (Malus × domestica)[22], and MtGSTF7 in Medicago truncatula[35], most of which belong to the Phi(F) class. Additionally, some plant ABC and MATE genes are involved in anthocyanin transport, including ZmMRP3 in maize[36], VvABCC1, VvAM1 and VvAM3 in grapevine[37,38], OsMRP15 in rice (Oryza sativa)[39], AtABCC2 and AtTT12 in A. thaliana[40,41], CaMATE1 in chickpea (Cicer arietinum)[42], SlMTP77 in tomato (Solanum lycopersicum)[43], and MtMATE2 in M. truncatula[44].
Roses (Rosa spp.) are ornamental plants with global economic importance. Some rose cultivars exhibit a post-anthesis transition from yellow to red/orange-red, such as the flowers of R. hybrida 'Masquerade'[45], R. hybrida 'Ehigasa' and 'Charleston'[46], R. hybrida 'Spectra'[47], and R. hybrida 'Chen Xi'[48]. The flower color change in 'Ehigasa' and 'Charleston' rose is attributed to the accumulation of anthocyanins, and the flowers of 'Chen Xi' and 'Spectra' do not turn red under shading conditions[46−48]. However, the mechanisms underlying post-anthesis color transition in roses are unclear. The butterfly rose (R. chinensis 'Mutabilis') is an ancient Chinese rose cultivar with single petals that change from light yellow to pink or dark pink during four-day anthesis. In this study, high-performance liquid chromatography-diode array detection (HPLC-DAD) and transcriptome analyses of butterfly rose samples were employed to determine the anthocyanin composition and molecular changes during PACC and identify the environmental factors influencing the PACC trait. This research offers a comprehensive analysis of the PACC trait in roses, as well as valuable information for understanding flower color evolution.
-
Samples of butterfly rose were collected from Kunming Yang Chinese Rose Gardening Co., Ltd., and planted in the germplasm garden of the Institute of Radiation Technology (116°43' N, 40°16' E) under open field conditions for 2–3 years. The PACC cultivar R. hybrida 'Spectra' was used for to verify candidate genes. Rosa hybrida 'Spectra' was cultivated in the China National Botanical Garden (North Garden). The collection of petal samples was authorized.
Different floral developmental stages of butterfly rose flowers, namely the bud stage (one day before anthesis, S3), first day of anthesis (D1), second day of anthesis (D2), third day of anthesis (D3), and fourth day of anthesis (senescent flowering stage, D4), were collected from the upper half of the petals between 08:00 and 09:00 on sunny days. More than 30 flowers were collected at each stage. Petal samples with additional anthocyanin coloration on the abaxial surface were excluded. The red part (post-change, SR) and yellow part (pre-change, SY) of the middle-layer petals of R. hybrida 'Spectra' were collected on the fourth or fifth day of anthesis in the morning (equivalent to the D2 stage of butterfly rose). The samples were collected from more than 15 R. hybrida 'Spectra' flowers. The fresh petal samples were cut, weighed, and immediately frozen in liquid nitrogen. Three biological replicates were collected, and samples were frozen and stored at −80 °C.
Sample treatments
-
Different treatments were used in this study to investigate the effects of light on butterfly rose petals, with buds about to open wrapped in different bags in the evening until the second day of anthesis (08:00–09:00) before flowering. First, paper bag treatment (PT) with semi-translucent paper bags (sketch tracing paper, Rotring, 78% light transmittance) was used under natural light–dark conditions. Second, aluminum foil bags were used for dark treatment (DT)[49]. Petals were sampled in the morning on the second day of anthesis. Additionally, in the afternoon (17:30) on the first day of anthesis, the flowers (equivalent to stage D1.5) were wrapped in aluminum foil bags until the next morning (08:00–09:00) (D1.5–2), and the petals of stages D1.5 and D1.5–2 were collected. More than 30 flowers were included under PT and DT, with more than 15 flowers collected from D1.5 and D1.5–2. D2 samples, which were opened under natural sunlight, were used as controls.
Measurement of anthocyanins
-
Anthocyanins were extracted according to the methods described by Wan et al.[50]. Approximately 0.1 g (fresh weight) of the sample was ground into a powder, transferred into a centrifuge tube, and extracted overnight at 4 °C using a 2 mL mixture of methanol:water:methane acid:trifluoroacetic acid (70:27:2:1, v/v/v/v). The extracts were centrifuged at 10,000× g and 4 °C for 10 min, and the supernatants were collected and filtrated (PTFE, 0.22 μm, Anpel). HPLC-DAD analysis was performed using an Agilent 1200/G1315D system in the wavelength range of 200–700 nm. The mobile phases comprised 0.5% aqueous formic acid (A) and acetonitrile (B). The gradient program has previously been described by Wan et al.[51]. A Zorbax SB-C18 analytical column (250 mm × 4.6 mm, 5 μm) was used. The column temperature, injection volume, and flow rate were set at 25 °C, 20 μL, and 0.5 mL/min, respectively. Anthocyanins were detected at a wavelength of 520 nm. In preliminary experiments, the anthocyanin composition was analyzed using ultra-performance liquid chromatography-electrospray tandem mass spectrometry (ExionLC AD; MS, Applied Biosystems 6500 Triple Quadrupole) using mixed petals at different developmental stages. Eight major anthocyanin components were identified. Anthocyanins were quantitatively analyzed using an external standard method[50]. Five anthocyanin standards, including cyanidin-3,5-O-diglucoside (Cy3G5G), cyanidin-3-O-glucoside (Cy3G), peonidin 3,5-O-diglucoside (Pn3G5G), peonidin 3-O-glucoside (Pn3G), and pelargonidin-3-O-diglucoside (Pg3G), were purchased from Sigma-Aldrich Chemical Co., Inc. (St. Louis, MO, USA). The remaining anthocyanins were quantified using a Cy3G standard curve.
Genes involved in anthocyanin biosynthesis in R. chinensis
-
According to the literature and our previous work, we identified 16 structural genes (excluding the UDP-glycosyltransferase family) involved in the flavonoid/anthocyanin biosynthetic pathway (Supplemental Table S1)[16]. The hidden Markov model profile PF00201 was used to search the rose genome for genes belonging to UDP glycosyltransferases (UGT), with an E-value of < 0.001, resulting in 217 candidate UGT genes[52]. Several transcription factor families involved in the regulation of anthocyanin biosynthesis have been identified in the rose genome, including 121 R2R3-MYB[53], 187 WD40[54], 100 bHLH[55], 48 basic leucine zipper (bZIP)[56],116 NAC[57], and 56 WRKY genes[58]. In addition, 23 BBX genes were re-identified in the rose genome based on a previous study[59].
Identification and analysis of GSTs in R. chinensis
-
The hidden Markov model profiles PF02798 (GST_N) and PF00043 (GST_C), downloaded from the Pfam website[60], were used to identify candidate GSTs in R. chinensis with an E-value of < 0.001. Additionally, 65 AtGST sequences (Supplemental Table S2) were downloaded from the genome of A. thaliana[61] and used as a query to search for candidate GSTs in the R. chinensis genome (
https://lipm-browsers.toulouse.inra.fr/pub/RchiOBHm-V2/ ) using the BLASTp program with an E-value cutoff of 1e−5. All non-redundant RcGST candidate genes were further verified by submission to the SMART website (http://smart.embl.de/ )[62]. The AtGST gene family contained 12 conserved motifs (E-value < 0.001), including GST_N (PF02798), GST_N_2 (PF13409), GST_N_3 (PF13417), GST_N_4 (PF17172), GST_C (PF00043), GST_C_2 (PF13410), GST_C_3 (PF14497), GST_C_6 (PF17171), EF1G (PF00647), Glutaredoxin (PF00462), Hemerythrin (PF01814), and membrane-associated proteins in eicosanoid and glutathione metabolism (MAPEG; PF01124) (Supplemental Fig. S1)[63]. Candidate RcGSTs and 65 AtGSTs were used to construct a neighbor-joining phylogenetic tree. RcGSTs in the same subfamily with the same domain(s) as A. thaliana were identified as full-length GST genes, whereas those with incomplete domain(s) were identified as partial GST (GST-p) genes (Supplemental Fig. S2).The identified full-length RcGST protein sequences were aligned using MAFFT 7.0 with default parameters[64], and phylogenetic trees were constructed using the maximum likelihood method on the RAxML online platform with 100 bootstrap replicates. The RcGST genes were mapped onto chromosomes based on the genome annotation document using TBtools, and tandemly duplicated genes were identified based on previous studies[65,66]. An intraspecies collinearity analysis of R. chinensis was conducted to identify segmentally duplicated genes. To study the evolution of GST genes among different plants, collinearity relationships were analyzed to infer the inter-species orthology between R. chinensis and other plants using TBtools software. Whole-genome sequences and annotation documents of peach, strawberry, and pear (Pyrus communis) were downloaded from the Genome Database for Rosaceae[67]. Whole-genome sequences and annotation documents of apple (Malus × domestica 'Golden Delicious') were downloaded from the official website[68]. Whole-genome sequences and annotation documents of grapevine, soybean (Glycine max), Medicago truncatula, and Populus trichocarpa were downloaded from the Ensembl website. An evolutionary tree between species was generated using the LifeMap website[69].
RNA-seq analysis
-
Twenty-one RNA-Seq libraries were generated from seven samples (S3, D1, D2, D3, D4, PT, and DT), and raw sequence data were deposited in the Genome Sequence Archive at the National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA006521)[65]. Fragments per kilobase million (FPKM) were used to determine gene expression.
PacBio Iso-Seq long-read sequencing
-
For PacBio Iso-Seq, full-length cDNA from D2 samples was constructed according to the method described by Liu et al[70]. The amplified cDNA products were built into SMRTbell template libraries in accordance with the IsoSeq protocol (PacBio). The SMRTbell template was then annealed to the sequencing primer and bound to the polymerase. Finally, the templates were sequenced on the PacBio platform by Biomarker Technologies (Biomarker Technologies, Beijing, China). SMRTlink v10.0 software was used for Iso-Seq data analysis. The open reading frames were detected using TransDecoder v5.0 for isoform sequences to obtain coding and untranslated region sequences.
Quantitative real-time PCR (RT-qPCR) analysis
-
Total RNA was isolated from different rose samples using an OmniPlant RNA Kit (DNase I) (CoWin Biosciences, Taizhou, China). cDNA was synthesized by reverse transcription from 15 μL of total RNA using the MonScript™ RTIII All-in-One Mix with dsDNase (Monad Biotechnology Co., Ltd, Wuhan, China). RhUBI2 (JK618216) was used as an internal control[71]. The primers used for real-time PCR are listed in Supplemental Table S3. Real-time PCR was performed as previously described[65].
Statistical analysis
-
Significant differences (p < 0.05) were determined using Student's t-test and one-way analysis of variance (ANOVA) tests. The t-test was performed using Microsoft Excel 2019 (Seattle, Washington, USA), and ANOVA was conducted using SPSS 23 (SPSS Inc., Chicago, IL, USA). Differential expression analysis between rose samples was performed using DeSeq2 in the OmicShare tool, with a Q-value threshold of 0.05. Venn diagrams were plotted using InteractiVenn[72]. A protein–protein interaction network was generated using the STRING database[73]. Correlation analysis was performed using the corrplot package[74].
-
In the natural environment, the petals of butterfly rose were light yellow at the bud stage (S3) and newly opened flowering stage (D1), turning pink on the second day of flowering (D2) then dark pink at the late flowering stage (D3 and D4) (Fig. 1a). The total anthocyanin content was low at S3 and D1 then increased significantly at D2 and later stages (D3 and D4), resulting in a color change from light yellow to pink and dark pink (Fig. 1b).
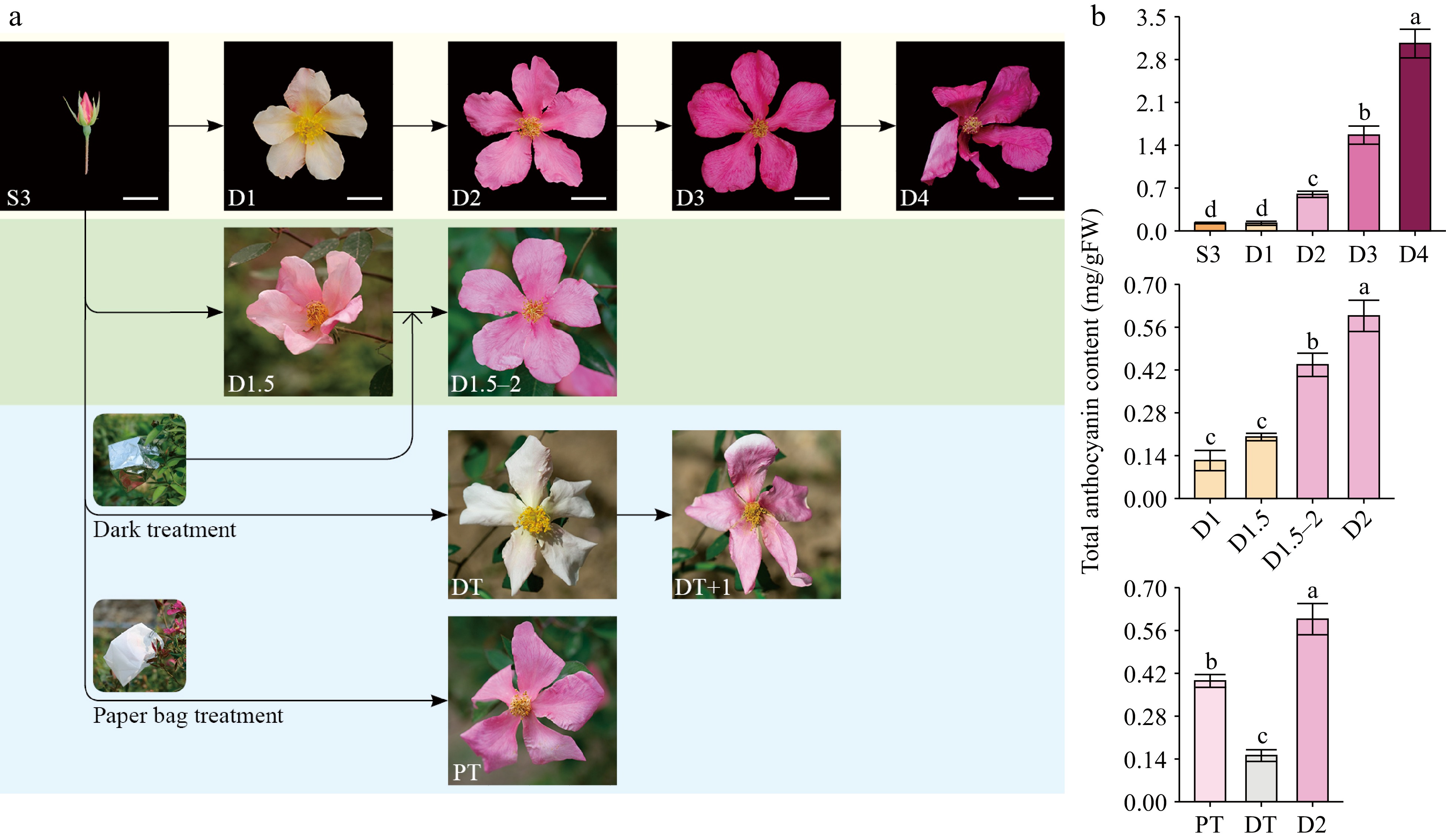
Figure 1.
Phenotypes and total anthocyanins contents of butterfly rose (R. chinensis 'Mutabilis') samples. (a) Photos of butterfly rose flowers under natural conditions and different treatments. Scale bars = 2 cm. S3, bud stage (one day before anthesis); D1, first day of anthesis; D2, second day of anthesis; D3, third day of anthesis; D4, fourth day of anthesis. PT, paper bag treatment, DT, dark treatment; DT+1, dark treatment flower exposed to natural sunlight conditions for one day. D1.5, flowers collected in the afternoon (17:30) on the first day of flowering; D1.5–2, flowers at the D1.5 stage that were dark treated until the next morning. (b) Total anthocyanin contents of different butterfly rose samples (n = 3–4). Different lowercase letters indicate statistically significant differences (ANOVA test, p < 0.05).
The butterfly rose petals turned light pink in the afternoon on the first day of anthesis (D1.5) and the total anthocyanin content of D1.5 samples was slightly higher than that of D1 samples. When D1.5 flowers were subjected to dark conditions until 08:00–09:00 the next day, the petals of D1.5–2 samples were pink and similar in color to those of D2 samples, but with a slightly lower total anthocyanin content (Fig. 1b). This may be because the D1.5–2 samples lacked 3–4 h of evening and morning sunlight. Thus, dark conditions (approximately 15 hours) had little effect on anthocyanin transport.
Treatments with different light intensities were used to study the effect of sunlight on flower color transition. Under PT, the petals of butterfly rose turned pink, similar to those of D2 samples under natural light conditions (Fig. 1a). Under DT, the petals turned almost white on the second day of anthesis. When DT flowers were exposed to natural sunlight for one day, the petal color changed back to pink (Fig. 1a). The total anthocyanin content in PT samples (reduced sunlight) was lower than that in D2 samples under natural light conditions, whereas DT samples (no sunlight) showed low accumulation of total anthocyanins (Fig. 1b). This suggests that sunlight was an important environmental factor influencing PACCs in butterfly rose flowers.
Anthocyanin contents in different samples of butterfly rose
-
Five butterfly rose samples at different developmental stages and two samples under different treatments were selected for anthocyanin analysis (Fig. 2a). Eight anthocyanins were detected in butterfly rose petals in the seven samples (Fig. 2b). More anthocyanin compounds were detected in dark pink samples (D3 and D4). Among the five developmental stages, the contents of five anthocyanins differed significantly between pink and light-yellow samples. Notably, Cy3G had the highest content in all pink samples, accounting for 72% of the total anthocyanin content at D2 and 86% at D4. Cy3G5G had the second-highest anthocyanin content. The other three differentially accumulated anthocyanins had very low contents (< 0.03 mg/gFW in D2 samples). Among the treated and control samples, five anthocyanins were differentially accumulated between DT and sunlight exposure (D2 and PT) samples, including the two main compounds, Cy3G and Cy3G5G (Fig. 2c). Therefore, the change in petal color from light yellow to pink in butterfly rose was caused by the accumulation of high anthocyanin contents, with Cy3G and Cy3G5G being the main components involved in petal coloration.
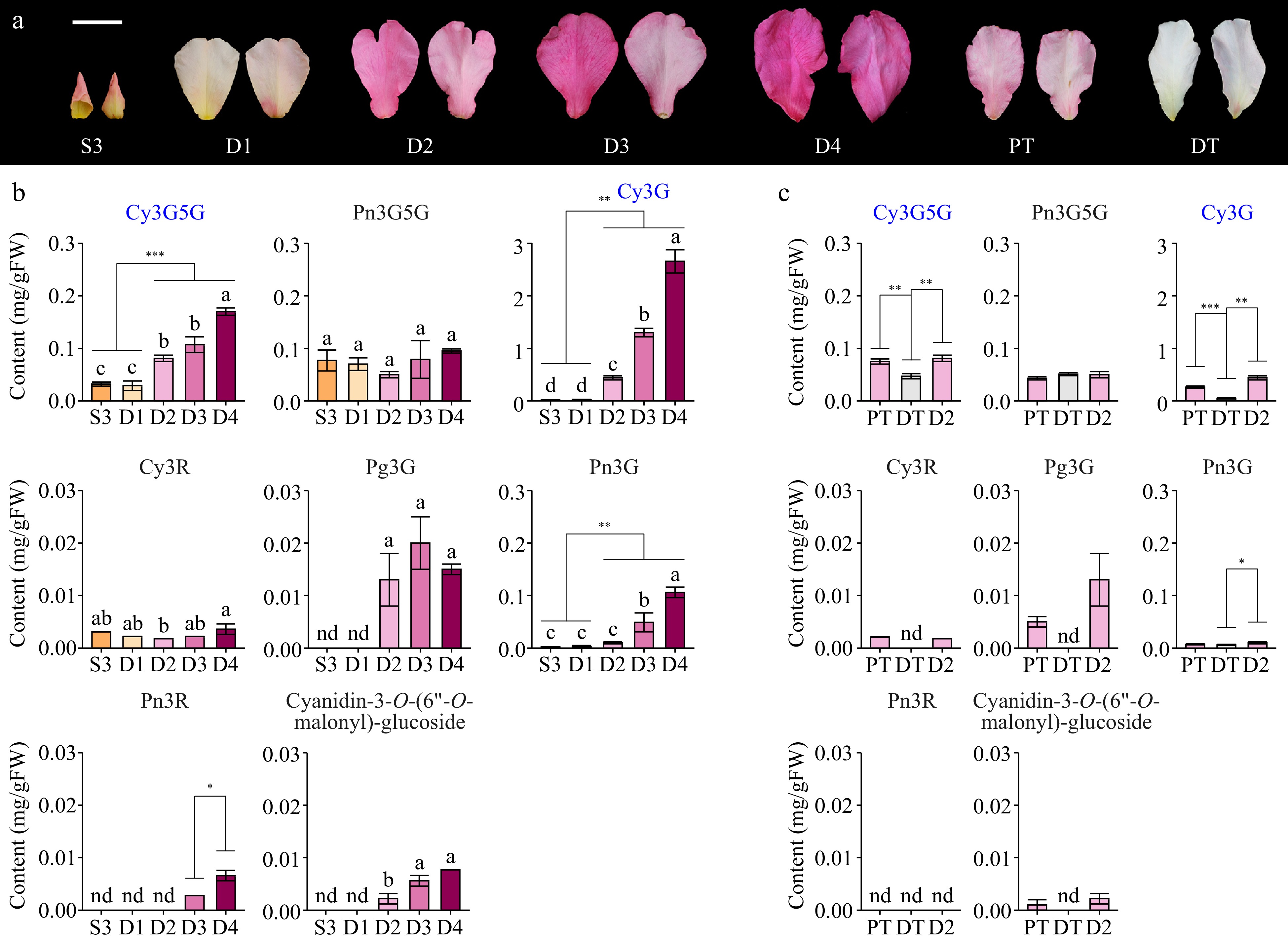
Figure 2.
Anthocyanin compositions in different butterfly rose samples. (a) Adaxial and abaxial surfaces of the petals are shown. Scale bar = 2 cm. (b) Anthocyanin contents of butterfly rose petals at different stages. (c) Anthocyanin contents of butterfly rose petals under different treatments. Data are presented as the mean ± standard error (n = 3–4). Pn3R, peonidin-3-O-rutinoside; Cy3R, cyanidin-3-O-rutinoside. Key anthocyanins are highlighted in blue. Different lowercase letters indicate statistically significant differences (ANOVA test, p < 0.05). nd, not detected. *p < 0.05, **p < 0.01, ***p < 0.001 by Student's t-test.
Structural genes involved in the anthocyanin biosynthetic pathway
-
Anthocyanins are biosynthesized via the flavonoid/anthocyanin biosynthetic pathway, and most structural genes involved in this pathway have been identified (Fig. 3a, Supplemental Table S1). Samples collected at different developmental stages (S3, D1, and D2) and under different treatments (D2, PT, and DT) were selected for further analyses. The expression patterns of structural genes involved in the anthocyanin biosynthetic pathway (except for UGT genes) in the different samples differed from those of key accumulated anthocyanins (Fig. 3b). Among the 217 candidate UGT genes obtained from the preliminary screening, only 91 were expressed in these five samples (average FPKM ≥ 1). Cluster analysis (Pearson correlation) was performed on the expression patterns of the 91 UGT genes and the contents of key anthocyanins (Fig. 3c). Thirteen UGT genes showed high expression in pink samples and low expression in bud and DT samples. However, only one of these genes had a high expression level, whereas the rest had relatively low expression levels (Fig. 3d). This gene is homologous to RhUF3GT1(AB292796)[10] and UF3GT (AB239786, partial)[75] and was named RcUF3GT1. RhUF3GT1 and UF3GT were cloned from the R. hybrida 'Ehigasa' and R. hybrida 'Charleston', respectively, both of which are PACC cultivars, and recombinant RhUF3GT1 expressed in yeast can catalyze the 3-glucosylation of anthocyanidins but not flavonols[10]. In butterfly rose the expression pattern of RcUF3GT1 corresponded to the increase in Cy3G content and RcUF3GT1 encodes a 466 amino acid protein in the D2 sample that shares 97.86% identity with the RhUF3GT1 protein (Supplemental Table S4), indicating that they have the same function. Therefore, among the structural genes involved in the anthocyanin biosynthesis pathway, only RcUF3GT1 may be involved in PACC.
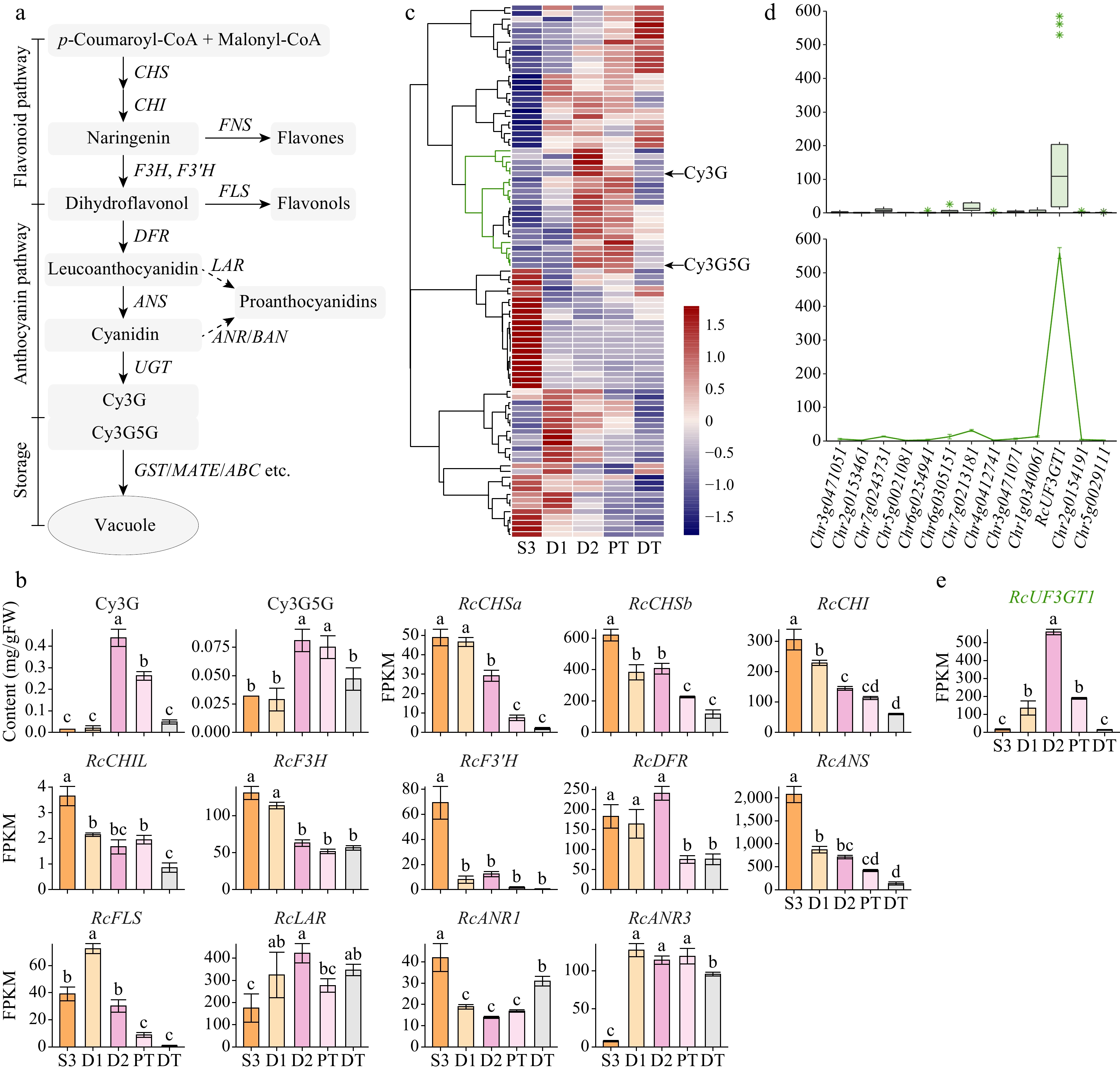
Figure 3.
Expression patterns of structural genes involved in the anthocyanin biosynthetic pathway. Data are presented as the mean ± standard error (n = 3). Different lowercase letters indicate statistically significant differences (ANOVA test, p < 0.05). (a) Schematic representation of the anthocyanin biosynthetic pathway in plant cells[23]. CHS, chalcone synthase; CHI, chalcone isomerase; FNS, flavone synthase; F3H, flavanone 3-hydroxylase; F3'H, flavonoid 3'-hydroxylase; FLS, flavonol synthase; DFR, dihydroflavonol reductase; ANS, anthocyanidin synthase; UGT, UDP-glycosyltransferase; LAR, leucoanthocyanidin reductase; ANR, anthocyanidin reductase; BAN, BANYULS. (b) Expression profiles of two key anthocyanins and structural genes in anthocyanin biosynthetic pathway (excluding UGT genes). (c) Clustering heatmap of UGT genes expressed in petals (Pearson correlation, clustering_method = 'complete'). (d) Expression levels of candidate UGT genes. Boxplot, average expression levels in five samples; line plot, expression levels in D2 samples. RcUF3GT1, RchiOBHmChr1g0383951. (e) Expression patterns of RcUF3GT1 in five samples.
Anthocyanin transport genes involved in post-anthesis color change
Identification and analysis of RcGST genes
-
In total, 80 full-length and 32 partial RcGST genes were identified (Supplemental Table S5). To explore the evolutionary relationships between RcGSTs, a maximum likelihood phylogenetic tree was constructed using 80 full-length RcGST proteins and 65 AtGST proteins (Fig. 4a). The RcGST family was divided into 12 classes: Tau (U), Phi (F), Lambda (L), Zeta (Z), Theta (T), tetrachlorohydroquinone dehalogenaselike (TCHQD), dehydroascorbate reductase (DHAR), γ-subunit of the eukaryotic translation elongation factor 1B (EF1Bγ), glutathionyl hydroquinone reductase (GHR), microsomal prostaglandin E synthase type 2 (mPGES-2), GSTs with two thioredoxins (GST2N), and MAPEG[61,76]. The GSTU class was the largest subfamily, with 50 full-length RcGST members, followed by the GSTF class, with seven. The other classes had fewer full-length RcGSTs, with no more than four in each class. Unlike Arabidopsis, no hemerythrin (GSTH) genes were detected in the rose genome. In addition, GST genes belonging to the metaxin class in the rose genome were incomplete (partial). Identified GST genes were renamed based on their subfamilies and chromosomal locations.
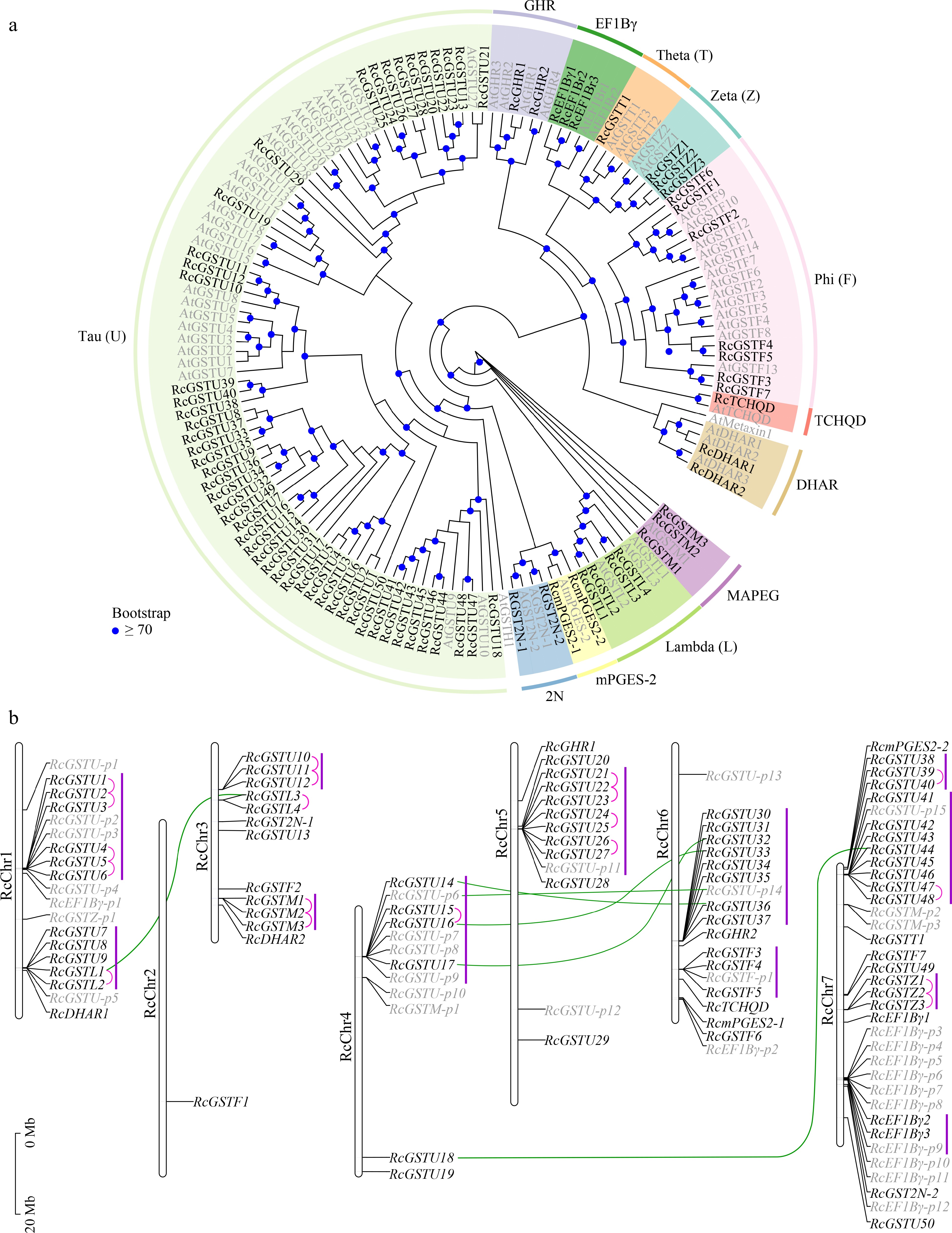
Figure 4.
Identification and analysis of GSTs in the R. chinensis genome. (a) Maximum likelihood phylogenetic analysis and classification of putative full-length RcGST genes. Genome IDs of AtGSTs (shown in gray) are listed in Supplemental Table S2. (b) Chromosomal distribution of full-length and partial RcGST genes. Full-length GST genes are represented by black letters; putative partial GST (GST-p) genes are represented by gray letters. Pink lines indicate tandemly duplicated genes; green lines indicate segmentally duplicated genes. Gene clusters are indicated by purple lines.
The mapping of 80 full-length and 32 partial RcGST genes to the chromosomes of R. chinensis revealed an uneven distribution among the chromosomes (Fig. 4b). RcChr 7 contained the largest number of GST genes, with 23 full-length and 13 partial RcGST genes, whereas only one RcGST gene was located in RcChr 2. Nineteen tandemly duplicated genes were detected in the R. chinensis genome, including 13 GSTU, two GSTL, two GSTZ, and two MAPEG genes. Additionally, six segmentally duplicated GST genes were detected, including five GSTU genes and one GSTL gene. These RcGST genes formed 12 gene clusters, including seven clusters comprising GSTU genes and one cluster comprising GSTU and GSTL genes. This indicates that the expansion of the GST gene family in R. chinensis was driven by tandem and segmental duplication, particularly in the GSTU and GSTL classes.
RcGST(s) involved in anthocyanin transport
-
Previous studies have shown a positive correlation between anthocyanin content and the expression of GST genes involved in anthocyanin transport[33,77]. To screen for GSTs involved in anthocyanin transport, we excluded genes with low expression levels (average FPKM < 1 in the five samples). A total of 42 full-length GST genes and five partial GST genes were expressed in these samples. Correlation analysis between gene expression levels and key anthocyanin contents showed that only RcGSTF2 and RcGSTU39 exhibited high correlation coefficients (Pearson r ≥ 0.80) with the two key anthocyanins (Fig. 5a). Analysis of the expression patterns indicated that only RcGSTF2 expression was consistent with the accumulation of key anthocyanins, particularly Cy3G (Fig. 5b). The amino acid sequences of 80 full-length RcGSTs were aligned with those of other functionally identified GST proteins using MAFFT software, and a neighbor-joining evolutionary tree was constructed using MEGA7.0 software (Fig. 5c). Phylogenetic analysis revealed that only RcGSTF2 clustered with the characterized GSTs involved in anthocyanin transport in other plants, especially other Rosaceae plants, indicating their functional similarity. These results indicated that RcGSTF2 is the only candidate GST gene involved in anthocyanin transport. To investigate the evolutionary relationship of GST genes involved in anthocyanin transport across different plants, rose and four other Rosaceae plants (peach, apple, pear, strawberry), two Fabaceae plants (Medicago truncatula and soybean), and one Salicaceae plant (Populus trichocarpa) were selected for inter-species collinearity analysis. These eight plants belong to the fabids of Rosales, with grapevine (belonging to Vitales) selected as the outgroup of the evolutionary tree. The results showed that GST genes involved in anthocyanin transport exhibited collinearity across these plants, indicating conserved evolution (Fig. 5d)[77]. The bioinformatics analysis showed that RcGSTF2 may be involved in the anthocyanin transport in butterfly rose.
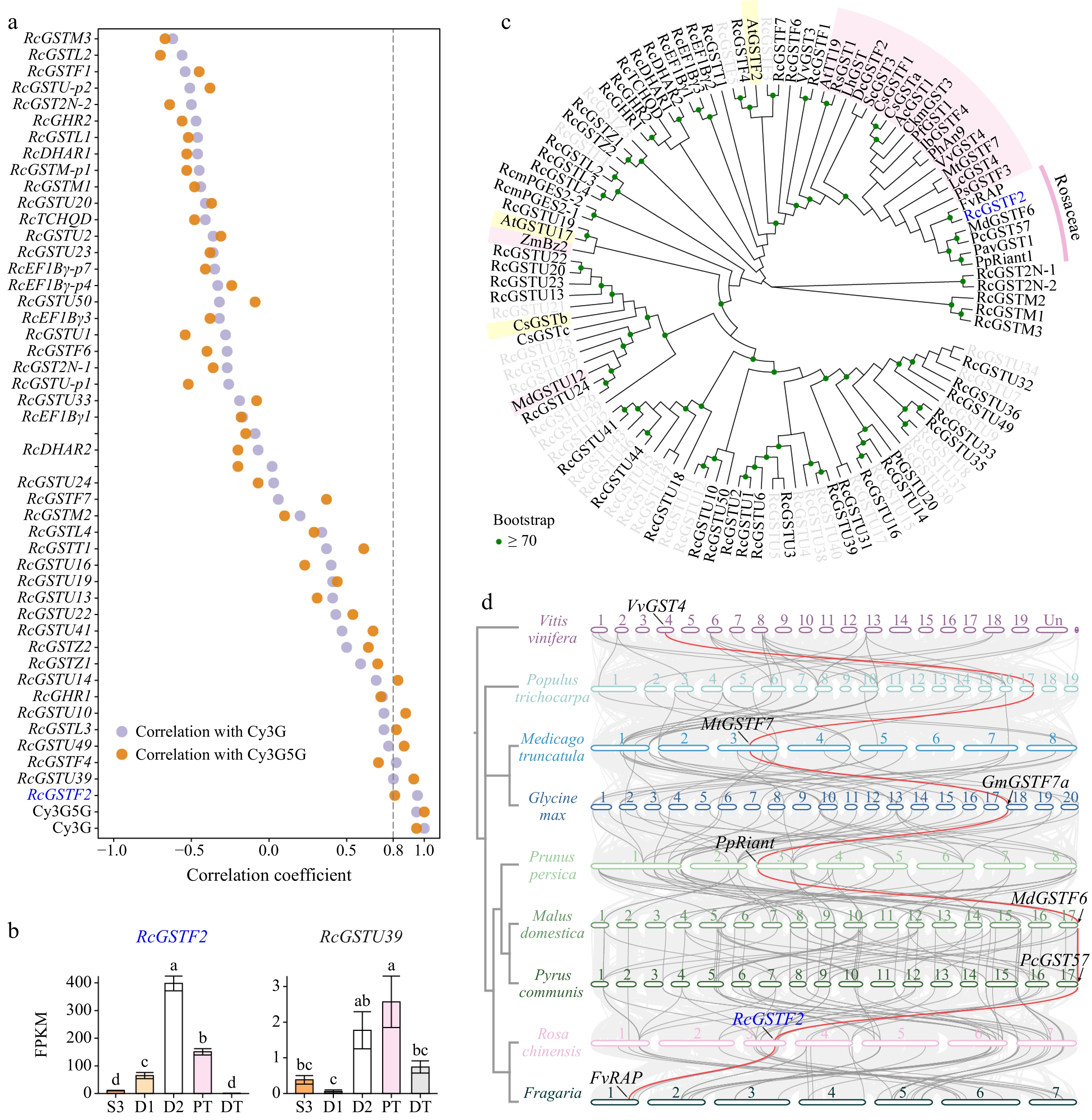
Figure 5.
GST(s) involved in anthocyanin transport. (a) Pearson correlation coefficient between petal-expressed RcGSTs and two key anthocyanins in five samples. (b) Expression profiles of two key anthocyanins and two candidate RcGSTs in different samples. Data are presented as the mean ± standard error (n = 3). Different lowercase letters indicate statistically significant differences (ANOVA test, p < 0.05). (c) Neighbor-joining phylogenetic tree of full-length RcGSTs and other characterized GST proteins. Encoding proteins of the RcGSTs that were not expressed in butterfly rose petals are represented by gray letters. GSTs involved in anthocyanin transport and flavonoid transport are highlighted with pink and yellow backgrounds, respectively. GST sequences used in this analysis are listed in Supplemental Table S6. (d) Inter-species collinearity analysis among eight fabid plants and grapevine. Collinear blocks are represented by light gray lines in the background; collinear GST genes are represented by gray lines; collinear genes of RcGSTF2 are highlighted in red. GSTs characterized in these species were labeled as follows: VvGST4, NP_001267869.1 (VIT_13s0067g03420); MtGSTF7, Medtr3g064700[35]; GmGSTF7a, Glyma.18G043700[35]; PpRiant (PpGST1), Prupe.3G013600[28,29]; MdGSTF6, MD17G1272100[22]; PcGST57, pycom17g27080[78]; FvRAP, FvH4_1g27460[32].
Other genes involved in anthocyanin transport
-
BLASTp analysis (pident ≥ 55%, E-value < 1e−50) identified nine orthologs of AtABCC2/VvABCC1/ZmMRP3/OsMRP15 in the rose genome. In addition, three orthologs of AtTT12/CaMATE1 (pident ≥ 70%, E-value < 1e−50) and seven orthologs of SlMTP77/MtMATE2/VvAM1/VvAM3 (pident ≥ 55%, E-value < 1e−50) were detected in the rose genome (Supplemental Table S7). Of these 19 orthologs, eight genes were petal-expressed (average FPKM in five samples ≥ 1). Most genes exhibited low correlation coefficients with Cy3G or Cy3G5G and only RcABCC2b exhibited a trend similar to that of anthocyanin accumulation (Fig. 6). However, only four partial transcripts of RcABCC2b were detected in D2 samples, and the identity of the longest protein sequence encoded by them and the corresponding genome protein sequence was 33.73% (Supplemental Table S4). Whether RcABCC2b was involved in the transport of anthocyanins in butterfly roses is not clear.
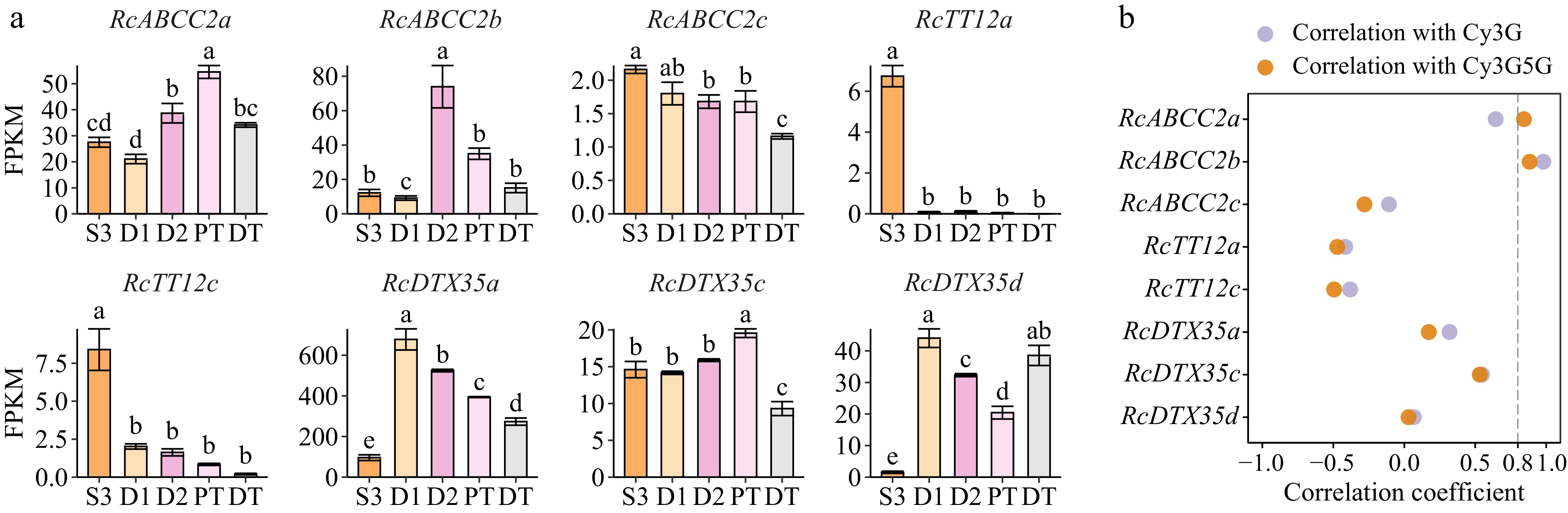
Figure 6.
Orthologs of anthocyanin-related MATE and ABCC transporters in butterfly rose petals. Their genome IDs are listed in Supplemental Table S7. (a) Expression patterns of ABC and MATE genes in different samples of butterfly rose. Data are presented as the mean ± standard error (n = 3). Different lowercase letters indicate statistically significant differences (ANOVA test, p < 0.05). (b) Pearson correlation coefficient between candidate genes and two key anthocyanins in five samples.
Other differentially expressed genes involved in post-anthesis color change
-
To identify other genes involved in the flower color change of butterfly rose, the following criteria were applied: (1) upregulated expression (Log2FC ≥ 1, Q < 0.05) in color-changing petals (D2) compared to bud (S3) samples, and (2) upregulated expression in pink samples (D2 and PT) compared to white samples (DT) (Fig. 7a). Venn analysis of the number of differentially expressed genes (DEGs) between different samples revealed that 754 genes were co-upregulated (Fig. 7b). Considering that the biosynthesis of anthocyanins in the D1 stage has just started, we calculated the correlation coefficients between the expression levels of these 754 candidate DEGs and the accumulation of key anthocyanins (Cy3G and Cy3G5G) for four samples (S3, D2, PT, and DT). Genes with higher expression levels (average FPKM ≥ 2 in four samples) and higher correlation coefficients (Pearson r ≥ 0.80) with both key anthocyanins (Fig. 7c) were selected.
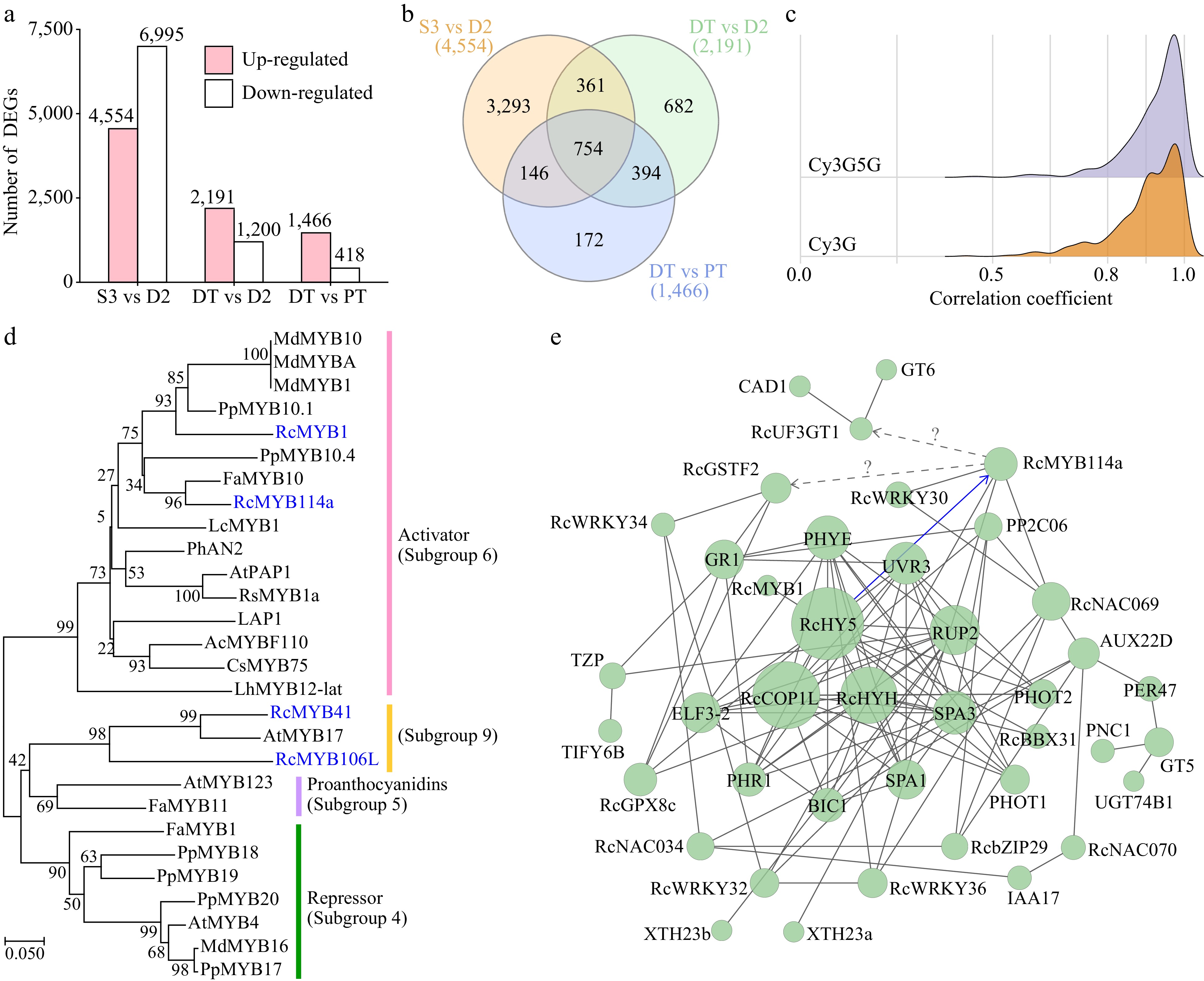
Figure 7.
Differentially expressed genes (DEGs) during the post-anthesis color transition. (a) Number of DEGs between different samples. (b) Venn analysis of the number of DEGs. (c) Correlation coefficients between DEGs (average FPKM ≥ 2 in four samples) and key anthocyanins. (d) Neighbor-joining phylogenetic tree of four candidate R2R3-MYB proteins (highlighted in blue) with other characterized R2R3-MYBs. Protein sequences used in this analysis are listed in Supplemental Table S9. (e) Protein–protein interaction network of DEGs (confidence = 0.20). Their genome IDs are listed in Supplemental Table S10. The identified interaction is displayed as a blue line.
A total of 445 DEGs were identified, including RcPAL2, 12 UGTs, RcGSTF2, and RcABCC2b. Among these, four R2R3-MYB genes (RcMYB1, RcMYB114a, RcMYB41, and RcMYB106L), four WD40 genes (RcSPA1, RcRUP1, RcCOP1L, and RcSPA3), three BBX genes (RcBBX28, RcBBX31, and RcBBX32), four bZIP genes (including RcHYH and RcHY5), three NAC genes, and four WRKY genes were identified as DEGs. No differentially expressed bHLH genes were detected. Key DEGs and their genome IDs are listed in Supplemental Table S8.
The protein sequences of the four differentially expressed R2R3-MYB genes, along with those of other R2R3-MYB protein sequences involved in anthocyanin biosynthesis, were used to construct a neighbor-joining phylogenetic tree (Fig. 7d). Among these, RcMYB41 and RcMYB106L belong to subgroup 9 and potentially involved in epidermal cell outgrowth[79]. RcMYB1 and RcMYB114a were clustered with MYBs that activate anthocyanin accumulation in other plants (subgroup 6) and positively regulate anthocyanin biosynthesis[17,18,80].
To predict the potential functions and relationships of these genes, we constructed a protein–protein interaction network among the DEGs involved in anthocyanin-related pathways. The results indicate that some BBX, bZIP, NAC, and WKRY transcription factors might be involved in PACC, while RcHY5, RcCOP1L and RcHYH might be the core regulators of color transition in rose petals. Indeed, RhHY5 induced the expression of RhMYB114a under light conditions in 'Burgundy Iceberg' rose[17]. However, the regulatory network of RcMYB114a, RcGSTF2 and RcUF3GT1 remain unclear.
Analysis of candidate genes
-
Based on the above analysis, 21 candidate DEGs including four structural genes (RcUF3GT1, RcGSTF2, RcABCC2b, and RcPAL2) and 17 transcription factors were selected for further analysis (Supplemental Table S4). The genes whose transcripts encoding protein sequences showed high identity (> 60%) with corresponding genome protein were selected for RT-qPCR analysis. The expression levels of 15 candidate genes were analyzed in butterfly rose and R. hybrida 'Spectra'), both of which were PACC cultivars (Fig. 8a). In R. hybrida 'Spectra' petals, no anthocyanins were detected in the yellow petal (pre-change, SY) samples, whereas more anthocyanins accumulated in the red petal (post-change, SR) samples (Fig. 8b & c). The main anthocyanins that accumulated in the SR samples were Pg3G (76.84%) and Pg3G5G (12.14%), which differed from the anthocyanin profiles of D2 samples.
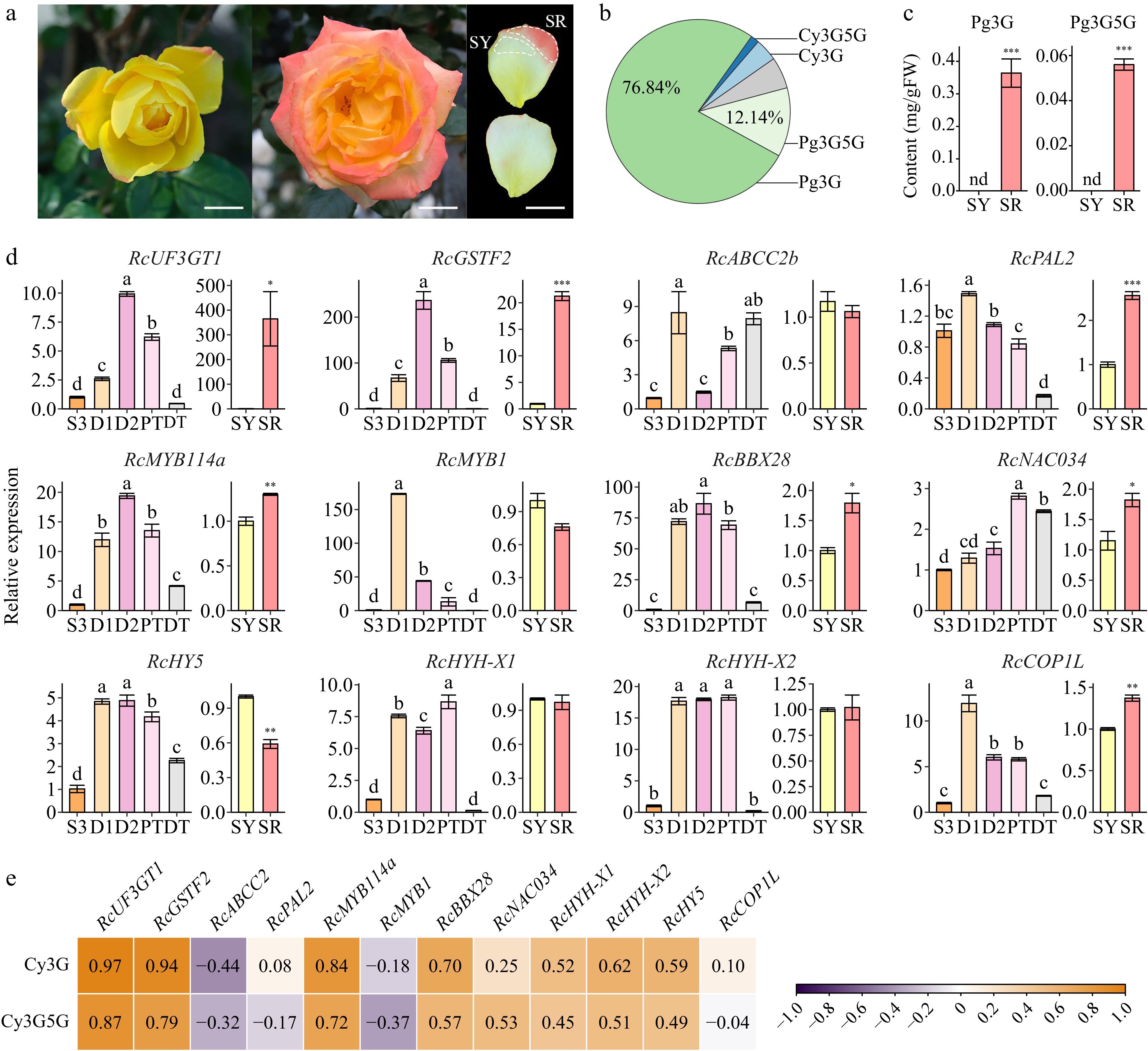
Figure 8.
Analysis of candidate DEGs. Data are presented as the mean ± standard error (n = 3). (a) Photos of R. hybrida 'Spectra'. Scale bars = 2 cm. Left, newly opened flower; middle, full-bloom flower (fourth or fifth day of anthesis); upper right, adaxial surface of collected petal; lower right, abaxial surface of collected petal. (b) Two key anthocyanin contents in different samples of R. hybrida 'Spectra'. (c) Anthocyanin components in the red part of middle-layer petals of R. hybrida 'Spectra' (SR). (d) Expression levels of candidate DEGs in different samples of R. chinensis (ANOVA test, p < 0.05). and R. hybrida 'Spectra' (Student's t-test, *p < 0.05, **p < 0.01). (e) Correlation analysis between the expression of candidate DEGs and key anthocyanins in butterfly rose samples.
In butterfly rose samples, RT-qPCR analysis confirmed the expression patterns of RcUF3GT1 and RcGSTF2, whose expression showed high correlations with anthocyanin contents, while the expression of RcPAL2 and RcABCC2b showed low correlations with anthocyanin contents. Similarly, RcUF3GT1 and RcGSTF2 were expressed higher in SR samples than in SY samples (Fig. 8d). In addition, RT-qPCR and correlation analysis on candidate transcription factors showed that RcMYB114a and RcBBX28 showed high correlations with anthocyanin contents in butterfly rose and were differentially expressed in R. hybrida 'Spectra' petals (Fig. 8d & e, Supplemental Fig. S3). Two distinct alternative splicing variants of RcHYH were detected in D2 samples (Supplemental Table S4), and the expression of RcHYH-X1 and RcHYH-X2 were more sensitive to sunlight than RcHY5. The expression patterns of RcHY5, RcHYH, and RcCOP1L were not strongly correlated with the anthocyanin content in different rose samples and their functions require further research. Together, these results suggest that RcUF3GT1, RcGSTF2, RcMYB114a and RcBBX28 are crucial genes involved in the post-anthesis transition in rose petals.
-
Flower color is a signal that plants use to communicate with their visitors; different messages can be sent to visitors by changing flower color. The retention of old flowers favors the attraction of visitors over long distances and directs nearby visitors toward rewarding flowers[3]. In this study, it was observed that visitors of butterfly rose flowers, including hoverflies and Italian bees, preferentially visited pre-changed (D1) flowers, followed by slightly post-changed (D2) flowers. White flowers after dark treatment were also popular among visitors. Hoverflies and bees preferred UV-absorbing yellow colors, which may contain pigments such as flavonoids and aurane chalcones[81]. From the bees' perspective, red appears similar to the background color of green leaves[3]. Therefore, plants such as butterfly rose change their flower color by accumulating anthocyanins in their petals to form different visual signals for visitors. This is a color-changing strategy used by many natural bee-pollinated plants[82].
PACCs in different plants are induced by different structural genes. The transition of flower color from acyanic (white and yellow) to cyanic (pink, red, and purple) is primarily due to an increase in anthocyanin content, accompanied by the upregulation of structural genes involved in the anthocyanin biosynthesis pathway[8,46,83]. In Viola cornuta, VcANS is the key regulated gene for floral color change during development, whereas the upregulated expression of NmCHS is associated with an increase in anthocyanin content in Nicotiana mutabilis petals[8,9]. In Pleroma raddianum, CHS and ANS are upregulated during color transition[84]. In the present study, among the structural genes involved in the flavonoid/anthocyanin biosynthetic pathway, only RcUF3GT1, and several other low-expression UGT genes showed similar expression patterns for anthocyanin accumulation in petals, similar to that in safflower (Carthamus tinctorius)[52]. Among previous studies on PACC rose cultivars, such as 'Charleston', 'Ehigasa', and 'Masquerade', all cultivars accumulate Cy3G and Cy3G5G in the post-change petals[45,46], which was the same for butterfly rose. Different anthocyanin profiles accumulated in the post-change petals of R. hybrida 'Spectra', mainly Pg3G and Pg3G5G (Fig. 8b). This suggests that anthocyanins causing the color change in rose were not limited to cyanidin glycosides. Therefore, we speculate that the glycosylation of anthocyanidin is regulated during the post-anthesis color transition in rose flowers.
Many rose cultivars are self-incompatible, such as R. chinensis var. spontanea and R. chinensis 'Slater's Crimson China'[85,86]. Under semi-transparent PT conditions, the exclusion of foreign pollen still resulted in a color change (Fig. 1a). This indicates that pollination may not be a key factor affecting PACC in butterfly rose. The same phenomenon was observed in Weigela japonica var. sinica, and its color change was independent of pollinator visits and flower pollination[2]. In the present study, a positive correlation was observed between anthocyanin accumulation and light intensity at the D2 stage. Butterfly rose petals showed minimal accumulation of anthocyanins under dark treatment but continued to accumulate anthocyanins after exposure to sunlight (Fig. 1a). It indicates that light is an important environmental factor affecting anthocyanin production.
In this study many differentially expressed transcription factors are involved in the light signaling pathway (Supplemental Table S10). As a major positive regulator of light signaling in plants, HY5 directly binds to the promoters of anthocyanin biosynthesis genes and MYB transcription factors to regulate anthocyanin synthesis[87,88]. A recent study on rose flowers showed that RhHY5 induces the expression of RhMYB114a and positively regulates anthocyanin biosynthesis by directly activating anthocyanin structural genes via the MYB114a-bHLH3-WD40 complex[17]. R2R3-MYBs can directly regulate structural genes involved in the anthocyanin biosynthesis, as well as GST transporter of anthocyanins[22,29]. Whether RcMYB114a can directly regulate RcUF3GT1 and RcGSTF2 requires further experimental verification. HYH also regulates anthocyanin accumulation in pear and peach[89,90]. In pear fruits, PybZIPa promotes anthocyanin biosynthesis by regulating PyMYB114, PyMYB10, PyBBX22 and PyUFGT[89]. In peach fruit, PpHYH activates PpMYB10 in the presence of the cofactor PpBBX4, leading to anthocyanin accumulation in sun-exposed peels[90]. PavBBX6 and PavBBX9 can positively regulate light-induced anthocyanin in Prunus avium by promoting PavUFGT, while the PpBBX16/PpHY5 complex strongly induced the promoter activity of PpMYB10 in Pyrus pyrifolia[19,91]. BBX28 negatively regulate flowering in Arabidopsis, and the PIF8-BBX28 module regulates petal senescence in rose flowers[49,92]. RcCOP1L does not interact physically with RcHY5, and its function is unknown[54]. Further research is required to elucidate the complex regulatory network involved in light-induced anthocyanin pigmentation in R. chinensis 'Mutabilis' flowers.
-
This study elucidated the mechanisms underlying color transitions in rose flowers. We found that color changes in butterfly rose flowers resulted from an increased accumulation of anthocyanins, with Cy3G and Cy3G5G being the key components. Trace amounts of anthocyanins accumulated in the dark-treated samples, whereas pigmentation occurred in the samples exposed to sunlight. Thus, sunlight plays a crucial role in the post-change pink coloration of R. chinensis 'Mutabilis'. Among the structural genes involved in the flavonoid/anthocyanin biosynthetic pathway, only RcUF3GT1 was significantly correlated with anthocyanin accumulation in butterfly rose flowers. Among the 80 genome-wide identified full-length RcGST genes, the expression patterns, and bioinformatics analyses highlighted the involvement of RcGSTF2 in anthocyanin transport. Orthologs of anthocyanin-related MATE and ABCC transporters were inactive in butterfly rose petals. RcMYB114a was considered an important positive transcription factor. Additionally, RcBBX28 might play significant roles in regulating anthocyanin biosynthesis during post-anthesis color change. These insights contribute to our knowledge of flower color change and have implications for further research on plant genetics and flower color evolution.
-
The authors confirm contribution to the paper as follows: study conception and design: Kong Y, Bai J; sample collection: Kong Y, Qiu L, Dou X, Lang L; laboratory analysis: Kong Y, Wang H; draft manuscript preparation: Kong Y; feedback on the analysis and manuscript: Wang H, Bai J. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
This research was funded by the Beijing Natural Science Foundation (6222007), National Natural Science Foundation of China (31401901), and the Innovation and Development Program of Beijing Academy of Science and Technology (23CB092).
-
The authors declare that they have no conflict of interest.
- Supplemental Table S1 List of structural genes involved in the anthocyanin biosynthesis pathway except the UDP‑glycosyltransferase (UGT) family in Rosa chinensis genome.
- Supplemental Table S2 Members of GST genes in Arabidopsis thaliana.
- Supplemental Table S3 Primer sequences of genes used for qRT-PCR analysis.
- Supplemental Table S4 The protein sequences encoded by transcripts of key genes.
- Supplemental Table S5 Members of GST and GST-p genes in Rosa chinensis and their sequence characteristics.
- Supplemental Table S6 Functionally identified GSTs in other plants.
- Supplemental Table S7 Other candidate genes involved in anthocyanin transport
- Supplemental Table S8 Key differential expressed genes during the post-anthesis color change in butterfly rose.
- Supplemental Table S9 Functionally identified R2R3-MYBs in other plants.
- Supplemental Table S10 Some differential expressed genes involved in anthocyanin-related pathway.
- Supplemental Fig. S1 Conserved domains of representative GST genes in Arabidopsis thaliana.
- Supplemental Fig. S2 Conserved domains of 80 full-length GST genes in Rosa chinensis genome.
- Supplemental Fig. S3 Expression patterns of four candidate transcription factors in different samples of Rosa hybrida 'Spectra'. Data are presented as the mean ± standard error (Student's t-test, *p < 0.05, ***p < 0.001).
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Kong Y, Wang H, Qiu L, Dou X, Lang L, et al. 2024. Anthocyanin contents and molecular changes in rose petals during the post-anthesis color transition. Ornamental Plant Research 4: e020 doi: 10.48130/opr-0024-0019 

Anthocyanin contents and molecular changes in rose petals during the post-anthesis color transition
- Received: 12 January 2024
- Revised: 15 May 2024
- Accepted: 21 May 2024
- Published online: 10 July 2024
Abstract: Flower color transitions during anthesis are taxonomically widespread; however, the mechanisms underlying post-anthesis color transition in roses are unclear. In this study, we collected petals of the butterfly rose (Rosa chinensis 'Mutabilis'), a post-anthesis color change cultivar, at different developmental stages and under different treatments. Anthocyanin composition and transcriptome data were analyzed to identify the environmental factors and crucial genes involved in post-anthesis color transition. The results showed that sunlight is a key factor triggering color transition. In butterfly rose flowers, color transition results from an increase in the accumulation of anthocyanins, primarily cyanidin-3-O-glucoside, and cyanidin-3,5-O-diglucoside. A combination of genome-wide identification, RNA-seq analysis, bioinformatics analysis, and quantitative real-time PCR verification revealed that RcUF3GT1 and RcGSTF2 genes were involved in anthocyanin production and anthocyanin transport, respectively. RcMYB114a may play a significant role in anthocyanin biosynthesis during color transition in roses, and RcBBX28 might be a crucial gene involved in this process. These insights contribute to our knowledge of flower color change and have implications for further research on plant genetics and flower color evolution.