









-
As an important epigenetic phenomenon, DNA methylation regulates the function of plant genomes without altering the DNA sequences; instead, various DNA methyltransferases are utilized[1,2]. Eukaryotic DNA methylation refers to the modification of the 5' cytosine in the CpG islands (CGI) sequence of the promoter, which reduces the binding ability of TF proteins to the DNA main helix groove, thereby inhibiting the transcriptional activity of genes[3]. In animals, 5-methyl-cytosine is observed at symmetric CpG dinucleotide sites, whereas plants exhibit methylation of cytosine in any DNA sequence environment, including symmetric CG and CHG and asymmetric CHH sites (H = A, T, or C); however, most sites are still dominated by methylated CG sites[2,3]. DNA methylation is essential for gene regulation, transposon silencing, and imprinting. DNA methylation, one of the most abundant epigenetic modifications in higher plants, predominantly occurs on transposons and other repetitive DNA elements, but plants use active demethylation to change the methylation state to regulate gene expression[4], for example, ROS1 mediates DNA demethylation pathways in Arabidopsis thaliana[5]. Although the production of specific patterns of DNA methylation is essential for many biological processes, exactly which key genes are regulated by DNA methylation, especially those involved in flowering induction, has not been elucidated.
Gene body methylation (gbM) refers to genes with enrichment of CG DNA methylation within the transcribed regions and depletion at the transcriptional start and termination sites. Gene body methylation (gbM) genes are enriched for housekeeping functions within angiosperm genomes[6]. Promoter-associated CpG island methylation status changes represent an important epigenetic regulation mechanism[7]. DNA methylation in the plant genome is more common in CpG islands, and these CpG islands are in the gene regulatory regions. The DNA methylation process is characterized by transcriptional inactivation and subsequent loss of function of the regulatory gene in this fashion without structural modifications[8]. CpG islands are generally observed at the same position relative to the transcription unit of equivalent genes in different species with some notable exceptions[3,9]. In mammals, CpG islands and CpG dinucleotides are 70 to 80% methylated at the 5' position of cytosine in the genome during DNA replication and transcription[10]. In Arabidopsis and rice, 80% of CpG islands are genetically related. CpG islands occur near the 5' end of the gene or cover the entire gene region. The location of the plant CpG islands in its related genes depends on the expression and organization of the gene. The degree of specific expression is related[11]. All these results suggest that CpG islands in plants may be useful means of deducing the expression pattern of uncharacterized genes[12].
The members of the methyl-CpG-binding domain (MBD) protein family play notable roles in the transcriptional regulation of genes and are important in determining the transcriptional state of the epigenome[13]. The MBD protein is an important recognition unit of methylation histology, and the machine produces the CpG site that requires the MBD domain and methyl-CpGs in the genome[14]. When a promoter recognition sequence of the transcription factor contains a methylated CpG site, a specific transcriptional inhibitor methyl-CpG binding protein (MBD) competes with the transcription factor for the methylation binding site. This competition leads to inhibition of the transcriptional expression of genes[15]. Based on the recognition of hypermethylated regions of the MBD7 protein, it also forms a complex with the IDM protein. The IDM protein recruits the ROS1 protein of the demethylase gene, thereby inhibiting the hypermethylation of genes and promoting their transcription[5].
MBD proteins serve as readers of the epigenome. Based on the ability of MBD proteins to bind methylated DNA in vitro, these proteins are primary candidates for reading DNA methylation[16−18]. Cytosine methylation is an important and extensive regulatory factor in plant systems, and high-throughput sequencing accelerates the characterization of methylation groups[19]. The MBD-seq methylation histology sequencing technique is based on the specificity of MBD proteins for recognizing highly methylated sequences[14,16,17]. MBD-seq is an effective and convenient method for observing differentially methylated regions[20]. This method has been widely used in humans and animals[20−25] and confirms the feasibility of high methylation region enrichment with the MBD2b protein in Arabidopsis[26]; in addition, it has not been used in plant research. Bisulfite sequencing results are the gold standard for DNA methylation detection, but this technique is difficult to perform on species with no reference genome. An effective and rapid method for detecting the methylation status of CpG islands region DNA is the use of methylation-specific PCR (MSP) technology[27,28].
The transition of flowering plants from vegetative to reproductive growth and the formation of floral organs play central roles in species development, reproduction, and evolution[29]. The flowering process of higher plants is achieved by integrating multiple internal and external flowering signals to regulate gene expression patterns[30]. The floral induction pathways primarily include the vernalization pathway, photoperiod pathway, autonomous pathway, gibberellic pathway, age pathway and T6P pathway[31]. The flowering regulation network is controlled by multiple genes. In addition to upstream and downstream interactions of multiple genes, there are a variety of apparent regulation mechanisms. Since epigenetic mechanisms play a key regulatory role in plant flowering signaling pathways[32], priority should be given to research on the epigenetic regulation mechanisms of flowering regulatory genes.
DNA methylation plays an important role in regulating the development of homologous transformation and flowering changes in plant organs[32]. For example, the epialleles of Arabidopsis thaliana, the FWA gene[33] and the SUPERMAN gene[34] all exhibit significant phenotypic variation. The laboratory treatment of C. lavandulifolium and cut chrysanthemum with 5-azaC produced flowering differences[35,36], and silent CmMET1 also acquired an early flowering phenotype[37]. All these results suggest that the methylation of some genes was inhibited during floral induction, but the genes regulated by DNA methylation have not been fully elucidated in C. morifolium or C. lavandulifolium.
Chrysanthemum (Chrysanthemum × morifolium Ramat.) is a well-known flower that originated in China. This flower is widely cultivated worldwide because of its extremely rich variety of flower shapes, flower colors and extensive ecological adaptability. Most Chrysanthemum species mainly flower in autumn. Chrysanthemum is a typical short-day plant that can be utilized as an important material for photoperiod research to study the molecular mechanism of photoperiod flowering.
C. lavandulifolium (Fisch.ex Trautv.) Ling et Shih is closely related to chrysanthemum, which is a diploid species widely distributed in China[38−40]. Considering the relatively simple genetic background, short growth cycle, and similarity to most varieties of chrysanthemum, it is an obligate short-day plant (SDP)[41] and is often employed as a model plant in biological studies[42−45]. The induction of flowering and development of capitulum in C. lavandulifolium were observed, and stages of development were classified[42,46]. The morphology and development of C. lavandulifolium were fitted to predict the developmental stages of inflorescences[46]. The transcriptome analysis technique was used to identify 56 important candidate genes that could be used for improvement in the chrysanthemum flowering period, which belong to the regulation of the photoperiod pathway, vernalization pathway, GA pathway, autonomous pathway, and FRI-dependent pathway[47]. The expression pattern and functions of key genes, such as DFL, members of the ClTFL, ClCRYs, ClPHY, CONSTANS-like gene family, and circadian clock genes, were studied, and all influenced the flowering of C. lavandulifolium. In particular, DFL, the homologue of the LFAFY gene, has been identified to promote flowering[41,45,48,49]. The chrysanthemum floral induction process involves many differentially expressed genes and differences in photoperiodic response mechanisms between different varieties which makes performing research on floral induction by DNA methylation in the process of regulatory mechanisms highly challenging[36]. Taking C. lavandulifolium as a model plant for scientific research on chrysanthemum was found to be an efficient method in our previous studies[35].
In view of the strong conservation of flower-related genes in angiosperms[50], we used the sequenced plant homologous genes and their promoter region CpG islands to predict the genes involved in flowering improvement. The possibility of DNA methylation regulation was evaluated, and the genes that may be regulated by DNA methylation in the process of floral induction of C. lavandulifolium were screened. On the other hand, the genes related to floral induction of C. lavandulifolium were obtained by the MBD-seq technique[20]. Therefore, we performed screening of DNA methylation regulatory genes involved in the process of floral induction of C. lavandulifolium while expression and DNA methylation were investigated by MSP[27] to verify the roles played by key genes in the floral induction process.
-
This study analyzed a total of 422 gene sequences, which are homologous genes of 42 flowering induction regulatory pathway genes, and found that 143 genes contained CpG islands. The CpG islands search results of each gene are shown in Supplemental Table S1.
These 42 genes contain four different floral induction pathway genes and six integron genes (Fig. 1a). The percentage of CpG islands varies among different homologous genes. For example, the percentages of ZTL, GAI, VIP2, and REF6 genes reached 100%, while some homologous genes (e.g., CCA1, LHY, and PIF3) have no distribution of CpG islands. The distribution of CpG islands has gene preference, and it is surmised that the DNA methylation regulation mechanisms of homologous genes may have certain similarities.
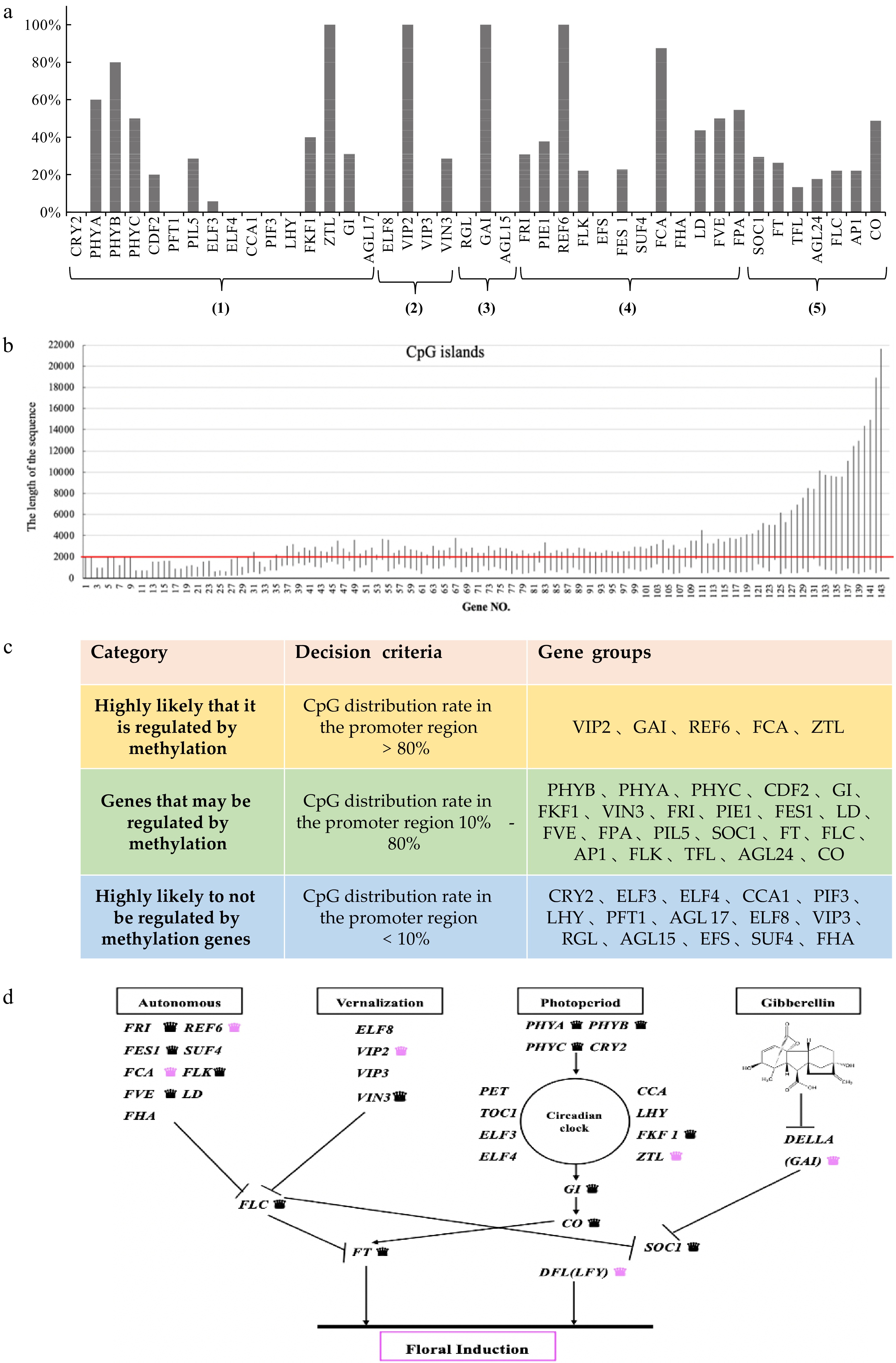
Figure 1.
Sequence analysis was used to analyse the flowering-related genes of C. lavandulifolium. (a) The percentage of CpG islands in the 42 homologous genes associated with flowering: (1) Photoperiod pathway; (2) Vernalization pathway; (3) GA pathway; (4) Autonomous pathway; (5) Floral pathway integrator. (b) CpG loci information of 143 genes in homologous genes. (c) Genetic classification based on the percentage of homologous CpG islands. (0−2,000 bp represents the promoter region). (d) Network of floral induction pathways in C. lavandulifolium (modified from Wang et al.[47]). Labelling of genes regulated by DNA methylation: black crowns refer to those that may be regulated by methylation genes, pink crowns refers to those highly likely regulated by methylation genes.
According to the CpG islands analysis of the information of all the sequences of a certain gene, all the genes can be finally divided into the following three categories which are associated with CG site methylation: highly likely to be regulated by methylation; possibly regulated by methylation; highly likely to not be regulated by methylation genes (Fig. 1c).
The results of screening for floral induction genes were placed in the floral induction pathways[47], and it was found that at least one of the four pathways is highly likely to be regulated by DNA methylation (Fig. 1d). The FCA and REF6 genes in the autonomous pathway, the ZTL gene in the photoperiod pathway, the VIP2 gene in the vernalization pathway and the GAI gene in the gibberellin pathway.
CpG islands location preference
-
We analyzed all gene sequences containing CpG islands. A total of 422 gene sequences of 42 homologous genes were analyzed, amongst them, 143 genes contained CpG islands which were mapped to find their locations. CpG islands were primarily distributed in the promoter region, and across the promoter region and gene body. The CpG islands mainly exist in the promoter region; in other words, the distribution of the CpG islands has a position preference (Fig. 1b).
MBD enrichment genome sequencing results
-
Box plots were performed on six samples of the two developmental stages of vegetative growth and budding, and the overall genomic DNA methylation level was characterized. The methylation levels of the six samples were largely the same and there was no significant difference (Fig. 2a).
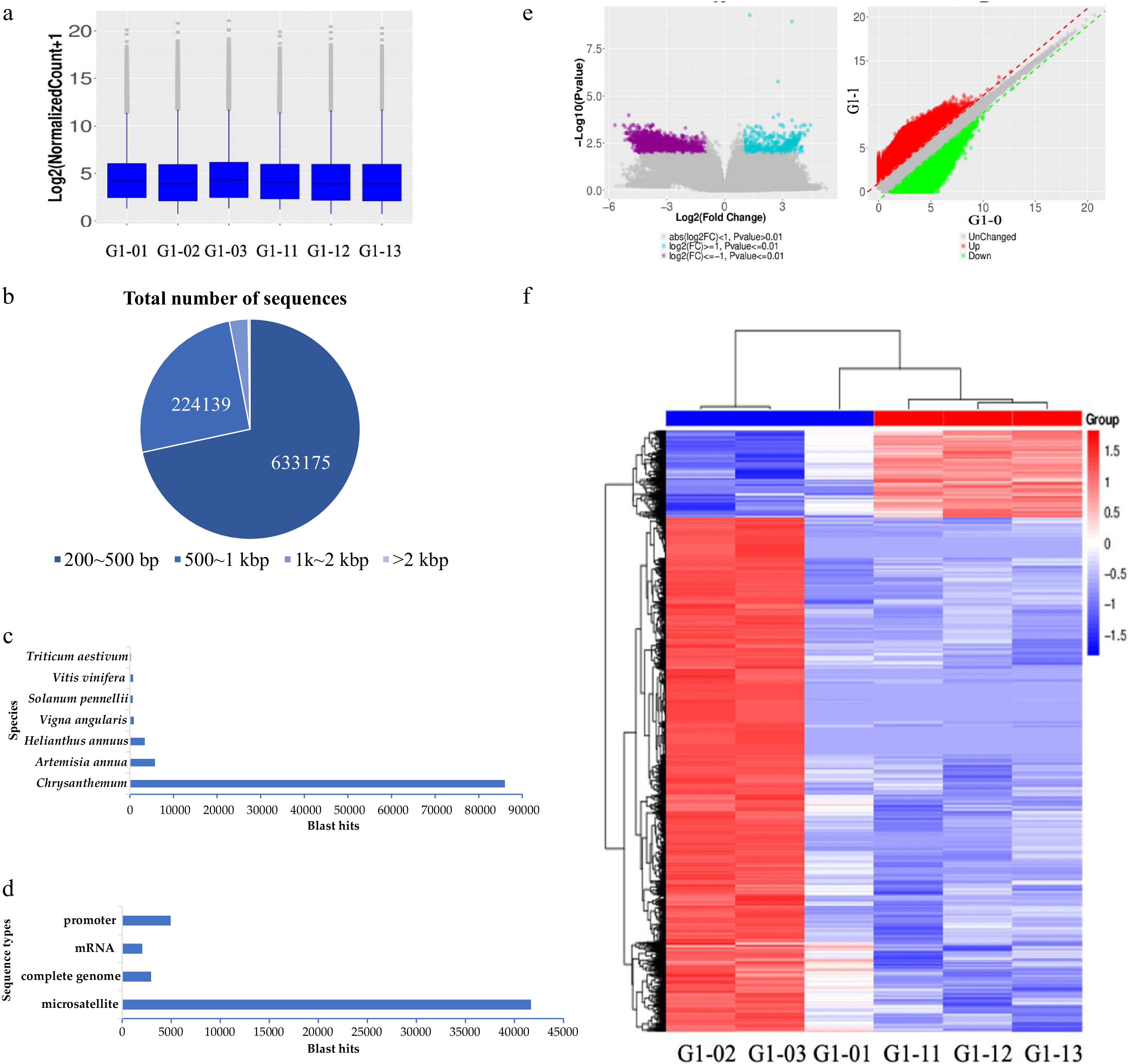
Figure 2.
MBD sequencing technology was used to screen the results of DNA methylated regulatory genes. (a) Boxplot of methylation levels for six samples. (b) The length distribution of nucleotides in MBD-seq sequencing results. (c) Nr annotation results in species statistics. (d) Nr annotation result sequence type statistics. (e, f) The volcano figure and heat map of differentially enriched genes between the seedling stage and bud stage.
The genomic MBD enrichment results of six samples of C. lavandulifolium were sequenced, and a total of 328,644,614 reads were obtained, which contained 430,941,997 nucleotide sequence information. The obtained DNA sequence was assembled by de novo sequencing to obtain 605,110 contig sequences. The CG content was 38.22%, the sequence length ranged from 224−28,176 bp, the average length was 312.81 bp and the N50 was 572 bp. Among these lengths, 200−500 bp, 500−1,000 bp, 1,000−2,000 bp and ≥ 2,000 bp each accounted for 71.63%, 25.35%, 2.74%, and 0.28%, respectively. The sequencing range was mainly between 200 bp and 500 bp, but there were also several long fragments (Fig. 2b).
Among these sequences, 118,289 sequences with good sequence quality were obtained and annotation by the NCBI Nr database was matched to 825 species (Fig. 2c). The sequence of Chrysanthemum was the most common, with a total of 85,973 cases accounting for 72.68%. Artemisia annua 5,755 cases accounted for 4.87%, Helianthus annuus 3,429 cases accounted for 2.90%, Vigna angularis 947, Solanum pennellii 685, Vitis vinifera 755 and Triticum aestivum 263. Of these species, 83.02% of the sequences were noted in the top seven species but mainly in the genus Chrysanthemum.
The sequence types of the annotated samples were analyzed, and the most common sequence types were microsatellites with a total of 41,675 sequences and highly repetitive sequences in plants were highly methylated (Fig. 2d). These sequence types were followed by the complete genome, usually the DNA sequence of the chloroplast or mitochondria. Our focus was on annotating the results for sequences of mRNA-coding and promoter regions.
Establishing the gene pool according to the results of the Nr database
-
The main gene annotations obtained from the Nr database were microsatellite sequences, complete sequences, mRNA sequences, promoter regions, and UTR regions. Nr is annotated and clustered, and similar structures or functions are summarized and counted for the cells with clear gene functions. A brief summary of gene names and functions provides an excellent gene pool for subsequent research, which can be divided into three main categories: genes that maintain basic metabolism; families of transcription factors; important single genes.
We performed a detailed analysis of the sequences of the promoter region and the mRNA region for the annotation results. In the Nr annotation results, there were 4,949 contig annotation results for promoter regions, having 3,488 sequences with unambiguous annotation results, which could be annotated as 28 genes. We summarize the results of the notes as shown in Supplemental Table S2.
The genes involved in floral induction include the ERF1 gene, the WRKY transcription factor and the LEAFY gene. Most of the remaining genes are enzyme genes in the secondary metabolic pathway, such as the diene synthase (AOC) gene, C4-sterol methyl oxidase gene, linalool synthase (LS) gene, and artemisinic aldehyde delta11(13) reductase (DBR2) gene.
In the Nr annotation results, there were 2044 contig annotation results for the mRNA region, with 137 sequences exhibiting clear annotation results. These results can be annotated as 61 genes. We summarize the results of the notes, as shown in Supplemental Table S3. Among the genes involved in floral induction are the DOF transcription factor family, WRKY transcription factor, GRAS protein, FT, PHYA, CRY 1a, GI, bHLH2, and AG1.
Differential enrichment sequence at the vegetative growth stage and bud stage
-
Through differential expression visualization, we can see the distribution of the differential enrichment sequence (Fig. 2e). In the vegetative growth stage and bud stage, the number of downregulated genes in the bud stage was less than the number of upregulated genes. This result indicates that the differential enrichment sequence has a significant change in DNA methylation status in several sequences during the flower development process, and that the overall methylation status of the genome has not changed significantly. Downregulated genes were more highly expressed than upregulated genes, indicating that DNA demethylation occurred with more genes involved in floral induction (Fig. 2f).
Differential enrichment analysis was performed between the two groups of samples at the vegetative growth stage and bud stage. According to the scatter plot, the sequences with significant upregulation and downregulation are shown. The sequences with significant differences produced 1738 annotation results. The set sequence was mainly microsatellite sequences, with a total of 1118 sequences, accounting for 64.32%, followed by mRNA sequences (81) and promoter regions (201).
The results of differential enrichment sequence annotation are in the promoter region, including the WRKY transcription factor, DBR2, ALDH1, AOC, CPR, LS, and the C4-sterol methyl oxidase gene, etc., as shown in Table 1.
Table 1. The result of enrichment of promoter difference sequence.
Contig No. Species Nr annotation 7 Artemisia annua aldehyde reductase (DBR2) gene 2 Artemisia annua ALDH1 gene 2 Artemisia annua allene oxide cyclase (AOC) gene 12 Artemisia annua artemisinic aldehyde delta11(13) reductase (DBR2) gene 140 Artemisia annua C4-sterol methyl oxidase gene 1 Artemisia annua amorpha-4,11-diene 12-hydroxylase 2 Artemisia annua cytochrome P450 reductase (CPR) gene 24 Artemisia annua epi-cedrol synthase gene 5 Artemisia annua linalool synthase (LS) gene 1 Artemisia annua WRKY-like transcription factor gene The result of the differential enrichment sequence was that the genes in the mRNA region included DELLA protein, DOF transcription factor, GRAS17, GRAS3, HB15, MYB44, MYB46, and S-adenosyl-L-homocysteine hydrolase, as shown in Table 2.
Table 2. Results for the enrichment of mRNA difference sequence.
Contig No. Species Nr annotation 1 Artemisia annua cytochrome P450 mono-oxygenase (cyp03) 1 Artemisia annua DELLA protein (DELLA) 1 Chrysanthemum × morifolium ChlH mRNA for magnesium chelatase subunit H 1 Chrysanthemum × morifolium DOF transcription factor 17 1 Chrysanthemum × morifolium GRAS protein (GRAS17) 1 Chrysanthemum x morifolium GRAS protein (GRAS3) mRNA, 1 Chrysanthemum × morifolium HD-ZIP protein (HB15) 1 Chrysanthemum × morifolium nitrate transporter 2.3 3 Chrysanthemum × morifolium trihelix protein (TH11) 6 Gymnocladus dioica succinate dehydrogenase subunit 4 (sdh4) 1 Helianthus annuus knotted-1-like protein 2 1 Medicago truncatula SPRY domain protein 1 Morus notabilis Calcium-transporting ATPase 2 1 Nicotiana tabacum S-adenosyl-L-homocysteine hydrolase (SAHH3) 1 Arachis duranensis tubulin alpha-4 chain 1 Beta vulgaris alanine--tRNA ligase 1 Beta vulgaris UDP-glucuronate 4-epimerase 6 1 Beta vulgaris zinc finger MYM-type protein 1-like 1 Brassica rapa 1-aminocyclopropane-1-carboxylate synthase 5 1 Brassica rapa condensin complex subunit 2 1 Camelina sativa L-ascorbate oxidase homolog 1 Capsicum annuum ABC transporter F family member 1 1 Capsicum annuum chaperone protein dnaJ 11 1 Capsicum annuum probable pectate lyase 8 1 Citrus sinensis F-box/kelch-repeat protein 1 Daucus carota E3 ubiquitin-protein ligase UPL1 1 Daucus carota ESCRT-related protein CHMP1B 1 Daucus carota inositol-tetrakisphosphate 1-kinase 1-like 1 Drosophila ficusphila glycine-rich cell wall structural protein 1.0 1 Erythranthe guttatus DNA topoisomerase 2 1 Fragaria vesca transcription factor MYB46 1 Gossypium hirsutum heat shock protein-like 1 Jatropha curcas ABC transporter C family member 4 1 Malus x domestica pectinesterase 3-like 1 Nicotiana sylvestris probable methyltransferase PMT11 1 Phoenix dactylifera MYB44 1 Ricinus communis ATP synthase subunit a 1 Sesamum indicum glycylpeptide N-tetradecanoyltransferase 1-like 1 Solanum tuberosum transcription elongation factor SPT5 Comparison of the results of two analytical methods
-
Based on the DNA hypermethylation status, we calculated the percentage of CpG islands in the promoter region of the homologous gene, and the results indicate that most of the genes might be regulated by DNA methylation (10%−80%). For example, 40% of the gene promoter region of the WRKY transcription factor, 62.29% of the homologous gene promoter region of the GRAS protein, 38.13% of the homologous gene promoter region of the DOF transcription factor family, 100% of the homologous gene promoter region of the LEAFY gene and 100% of the homologous gene promoter region of the DELLA protein were identified (Table 3). In early studies, it was found that the CpG island of the LEAFY gene promoter region and the demethylation of the LEAFY gene CpG island region are involved in the regulation of LEAFY expression during development[51], as determined by our research. The LEAFY gene was identified as a gene that is highly likely to be regulated by DNA methylation.
Table 3. Summary and analysis of key candidate genes in C. lavandulifolium.
Genes Type of methylation analysis CpG islands Promoter mRNA Enrichment of differences WRKY transcription factors family 40.00% Yes Yes Yes GRAS protein family 61.29% − Yes Yes DOF transcription factors family 38.13% − Yes Yes DELLA protein family 100.00% Yes Yes LEAFY 100.00% Yes − − FT 26.32% Yes − GI 31.03% Yes − PHYA 60.00% Yes − The sequence analysis technique is based on the possibility that sequence specificity may be regulated by DNA methylation. The MBD protein enrichment technique was used to detect the methylation status of the sequence. The genes we selected based on the two characteristics have certain mutual conformation characteristics. As DNA methylation plays a key role in floral induction, for the gene pool, further confirmation of its role requires validation through experimental analysis.
Expression analysis of candidate genes by RT-PCR
-
During the seven days of short daylight induction, almost all the detected genes were differentially expressed, the expression timing or pattern was different among all the genes. For example, the ClLS gene was expressed only on D3 (day 3), ClFCA was expressed only on D3 and D4 after flowering, and ClFLC, ClCOL4, ClGAI and ClMET were expressed from D3-D5. ClDOF was only expressed between D2 and D5. ClROS1 is expressed on D0, D4 and D5 (Fig. 3a).
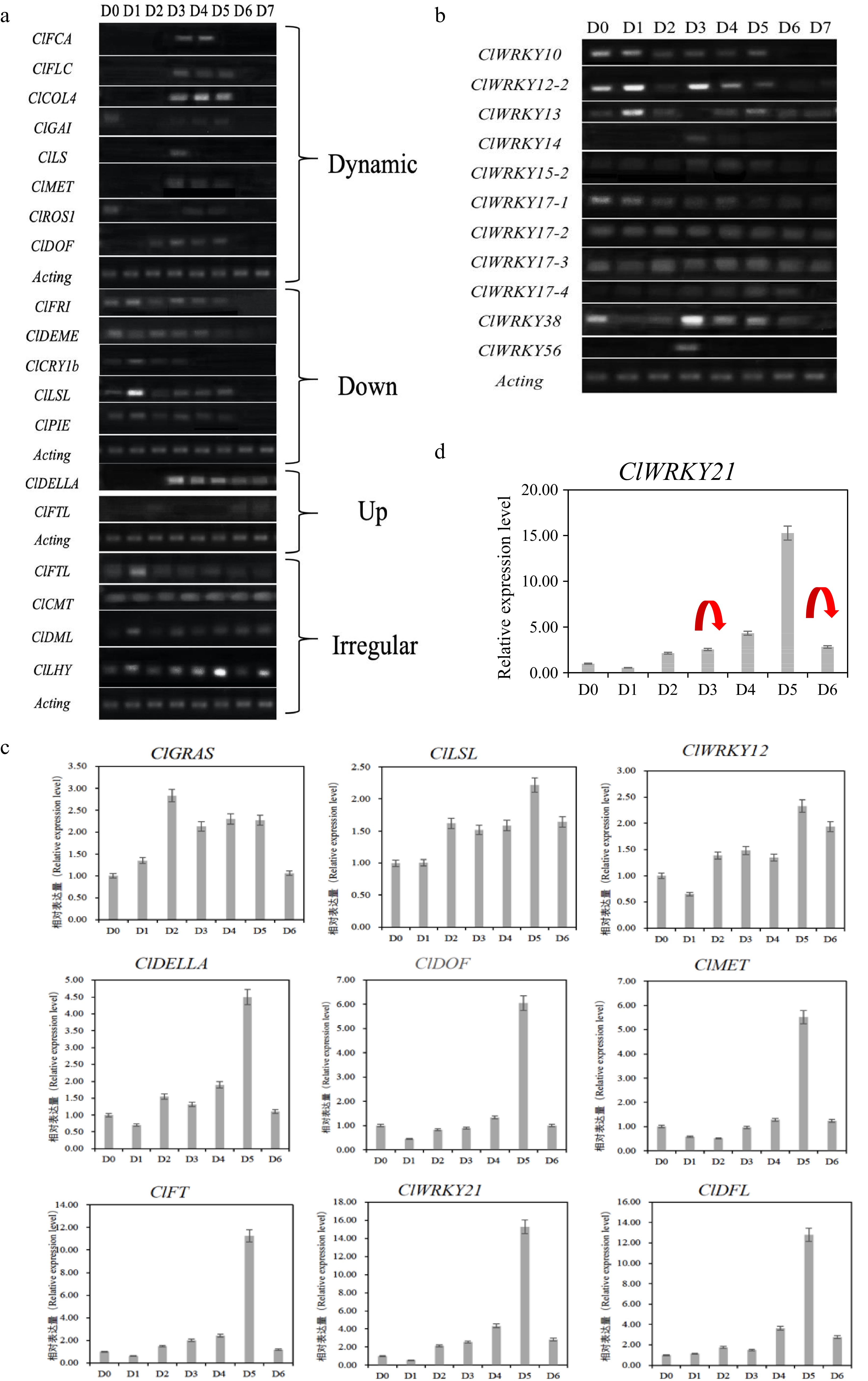
Figure 3.
The expression result of candidate genes in the floral induction process of Chrysanthemum lavandulifolium. (a) Dynamic changes of differentially expressed genes. (b) Differential expression screening of the WRKY gene family by RT-PCR. (c) Relative gene expression during floral induction of C. lavandulifolium. (d) Gene expression and DNA methylation status markers of ClWRKY21, the red arrows represent the disappearance of DNA methylation at this stage.
ClDELLA and ClFTL genes were not expressed at the initial stage of floral induction and were highly expressed at the subsequent stage. The DELLA protein gene was not expressed from D0 to D2 while highly expressed between D3 and D7, indicating that the DELLA protein played an important role during D3 to D7 to induce flower formation. The ClFTL gene was only expressed on D6 and D7 after two days (Fig. 3a). The expression of the ClFRI, ClDEMETER, ClCRY1b, ClLSL and ClPIE genes was downregulated in the floral induction process; in other words, they were highly expressed in the early stage of floral induction and not in the late stage of SD induction. ClFTL, ClCMT, ClDML, ClLHY and other genes showed no significant difference in expression during the induction period of short sunlight (Fig. 3a).
In addition, the expression of WRKY gene family members was analyzed by RT-PCR, among them, ClWRKY14, ClWRKY56, ClWRKY21-4 and ClWRKY15-2 were only expressed in the middle stage of floral induction, and ClWRKY10 showed downregulated expression, while ClWRKY12-2 and ClWRKY13 showed no obvious pattern (Fig. 3b). The WRKY gene family presents different expression rules in the floral induction process. It is possible that different members play different roles in the flowering induction process.
We performed a class quantitative analysis of the gene expression of 30 genes, including five DNA methylation-related genes, 14 flowering genes and 11 members of the WRKY gene family. There were differences before and after the floral induction of differentially expressed genes, and the expression pattern was different during the floral induction period. Different genes showed high expression at various stages of floral induction (early, mid, late), while some genes exhibit high expression in the floral induction period with no obvious differences. Five DNA methylation related genes (ClMET, ClDEMETER, ClCMT, ClDML, and ClROS1) were differentially expressed, indicating that DNA methylation plays an important role in the flowering induction process of C. lavandulifolium. Different members of the WRKY gene family also showed similar rules in the floral induction of C. lavandulifolium. Considering that it takes response time for DNA methylation to play a role in flowering, more detailed studies should be carried out on the genes with high expression in floral induction.
qRT-PCR expression analysis of candidate flowering genes
-
qRT-PCR was used to analyze the expression of some key candidate genes, and all nine genes showed a trend of differential expression in the flowering induction process, which increased earlier and then decreased in the later stages. Three of the genes (ClGRAS, ClLSL, ClWRKY12) had similar dynamic expression changes and the remaining six genes showed very similar expression trends in the induction of flowering, and their expression peaks all appeared at D5 (Fig. 3c).
In the early stage of induction, the expression of these six genes gradually increased, and the expression of the gene reached its peak at day 5 (the middle and late stages of floral induction) after induction and decreased sharply after the induction of flowering (late stages of flowering induction). Such genes play an important role in floral induction and flower development and are likely to be key regulatory factors in the floral induction pathway of C. lavandulifolium. The regulatory mechanism of DNA methylation in this gene should be further explored and studied.
Candidate gene promoter region methylation analysis (MSP)
-
After the sample was treated with bisulfite, if the fragment was amplified by a methylation-specific prime-M, the detected site will be considered methylated, On the other hand, if Primer-U amplify the fragment, that means the detected site did not show methylation. For example, ClWRKY21 was amplified only with nonmethylated specific primers, indicating the lack of DNA methylation. If both are amplified, two states, ClFT, DFL, and ClMET coexist across the genome. As the amplification results of the methylated fragments change during floral induction, it indicates that the methylated state of ClWRKY17 and ClWRKY21 has a dynamic change (Table 4).
Table 4. MSP results and DNA methylation status analysis.
Type Gene name Flowering induction stage Conclusion D0 D1 D2 D3 D4 D5 D6 Quantitative change ClFT-M + + + + + + + Both ClFT- U + + + + + + + DFL-M + + + + + + + Both DFL- U + + + + + + + ClMET-M + + + + + + + Both ClMET- U + + + + + + + Toqualitative change ClWRKY12-M × × × × × × × Unmethylation ClWRKY12- U + + + + + + + ClWRKY17-M + + + + + × + Dynamic change ClWRKY17- U + + + + + + + ClWRKY21-M + + × + + + × Dynamic change ClWRKY21- U + + + + + + + According to the results, the methylated state fragments of ClFT, DFL and ClMET coexist with the nonmethylated state; in other words, only the methylation quantity changes in the floral induction process, meaning that further quantitative analysis is needed.
ClWRKY21 showed a non-methylated state throughout the floral induction process, and the methylated state did not change. The methylation states of ClWRKY17 and ClWRKY21 changed during the floral induction process. The methylation state of ClWRKY17 changed at D5, while that of ClWRKY21 changed twice at D2 and D6. ClWRKY17 and ClWRKY21 were demethylated during floral induction, leading to high gene expression (Table 4).
In combination with the expression of the ClWRKY21 gene, the methylation state of the ClWRKY21 gene may have changed twice during the floral induction process (Fig. 3d).
When the expression level of the ClFT gene was changed, the methylation level of the ClFT gene was stable within a certain range without significant change. The DFL gene maintained the methylation level across all stages, however, it was higher before short day exposure and lower after short day exposure. The DFL gene may regulate gene expression through changes in DNA methylation levels (Fig. 4a). There is no corresponding rule between the expression of the ClMET gene and its methylation level, and the change in the ClMET gene expression level may be unrelated to the change in the DNA methylation level. The ClMET gene is an important DNA methylation transferase gene (Fig. 4a). Although this gene is not regulated by DNA methylation during floral induction, the difference in its expression level is likely to regulate gene methylation and achieve the ultimate goal of flowering regulation[37]. However, there is no clear evidence of the relationship between MET and DFL genes, and the high expression of MET may regulate other flowering inhibitors.
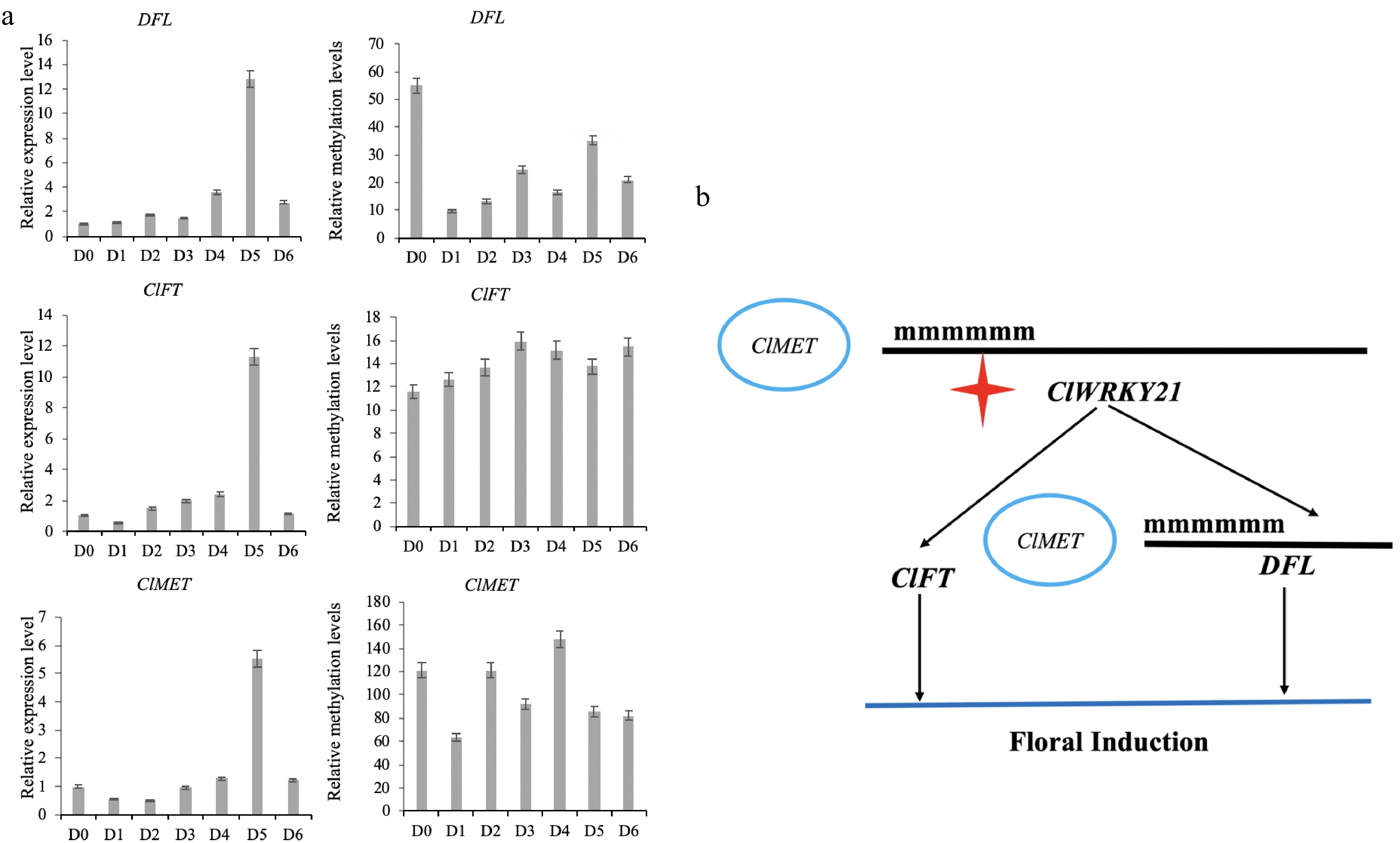
Figure 4.
The result of DNA methylation of key genes and construction of a floral induction network of C. lavandulifolium. (a) The expression level and methylation level of key flowering genes. (b) Role of the ClWRKY21 gene in floral induction of C. lavandulifolium.
Role sites of the ClWRKY21 floral induction regulation network
-
Based on the above results, the ClMET, ClWRKY21, DFL and ClFT genes, all play a key role in the floral induction of C. lavandulifolium. Specifically, these genes are differentially expressed in the floral induction, and D5 showed highest expression in the critical stage. There was no significant change in the DNA methylation level in the ClFT promoter region during floral induction, while the DNA methylation level in the DFL gene promoter region decreased gradually during short-day induced flowering (Fig. 4a). The promoter region of ClWRKY21 disappeared twice in the flowering process, suggesting that it is a key factor regulated by DNA methylation in the floral induction of C. lavandulifolium in response to short days. The dynamic changes in DNA methylation of the above genes may be affected by the expression level of the DNA methyltransferase gene ClMET (Fig. 4b). But ClMET is a flowering suppressor, and early flowering can be achieved by silencing the gene. In this study, the ClMET expression can only indicate that the gene is highly expressed in the flowering induction stage, and further research is needed on the downstream target gene of ClMET.
-
Many phenotypes are related to DNA methylation[5,33,34,52], and relevant studies on chrysanthemum are mostly related to flower characteristics. After the treatment of cut chrysanthemum and C. lavandulifolium with 5-azaC, we found that the treated materials showed abundant phenotypic variation[35,36], especially regarding the difference in flowering period, which was the most common. It was surmised that DNA methylation of some genes in the flowering process was inhibited, but the controlled genes were unknown. Changes in genomic methylation levels can only be detected by HPLC[53]. The result of differential methylation bands obtained by MSAP technology were not satisfactory[54−58]. Currently, the research in the model plant regarding plant flowering regulation networks has already been clearly determined, and key gene methylation levels can be targeted for testing, but because floral induction pathways are involved in many genes, how to determine which gene is expressed due to DNA methylation regulation is challenging. The screening of genes regulated by DNA methylation during the flowering process provides a reliable method for the study of DNA methylation of other genes to obtain candidate genes.
Exploration and screening of flowering genes in plants
-
Flowering is an important process for plants as they transform from the vegetative to reproductive stage[59]. During the flowering process of A. thaliana, the expression levels of about 2,000 genes are changed[60], amongst them, 80 genes are directly involved[61], engaging five major floral induction pathways[62]. In the transcriptome study of C. lavandulifolium, there were 14406 differentially expressed unigenes in the young and flowering stages and 211 flowering related unigenes were annotated in the transcriptome, of which 58 homologous genes were considered important genes for regulating anthesis[47]. Different expression levels of many genes are involved in the process of floral induction, which present challenges to the process of screening the genes regulated by DNA methylation in the process of floral induction.
We used sequence analysis to obtain genes that may be regulated by DNA methylation in the floral induction pathway, greatly narrowing the range of candidate genes and focusing on epigenetic regulation of genes. Next, on this basis, MBD-seq was used to supplement candidate genes, such as WRKY genes and GRAS proteins. Genes identified by both methods also exist, such as FT, PHYA, and CRY1a. Finally, according to the characteristics of the two methods, sequence analysis technology can be used to screen known genes, while MBD-seq technology is used as a method for unknown gene mining and DNA methylation differential gene screening.
Advantages and limitations of MBD-seq technology
-
In this study, the MBD protein was used to identify the specific characteristics of hypermethylated state sequences in plants, and sequence information were based on the perspective of the methylated state. Since highly repeated sequences in plant genomes are generally in a highly methylated state, microsatellite sequences account for a large proportion of the annotation results of enrichment sequences obtained in this study. We focus on the mRNA and promoter region sequencing, though we have obtained a few possible candidate genes, the efficiency was not satisfactory. If there is a corresponding reference genome, the annotation of protein enrichment results will be more detailed and accurate. The application of MBD-seq technology in plants is still in the exploration stage and only preliminary results have been obtained in this study. If combined with genomic sequencing information, a more effective technique to obtain differentially methylated regions can be developed. Although MBD-seq has limitations in its application in species without a reference genome, our results also showed that the method can still obtain desired results to a certain extent, which also provides a reference for related studies of other species without a reference genome.
Prospects of studies on genes controlling DNA methylation in flowering genes
-
In the field of medicine, through a comprehensive analysis of differential expression (DEGs) and differentially methylated regions (DMGs), genes that show both differential expression and methylation have been identified[63]. Finally, genes in the promoter region where hypermethylation is closely related to disease presentation are obtained. Based on mature gene-editing technology, several scholars have developed a demethylase editing tool based on CRISPR-dcas9, which contains the catalytic domain (CD) fusion of TET gene dioxygenase 1 (TET1-CD) and inactivated Cas9 (dCas9). Under the guidance of the designed single-directed RNA (sgRNA), the fusion protein can selectively demethylate the target region in the BRCA1 promoter, leading to upregulation of gene transcription and achieving targeted cytosine demethylation in mammalian cells[64]. Scholars have also developed related targeted DNA methylation editing systems (scFv-TET1 and dcas9-TET1)[65−68] and are widely used in animal research, but research on targeted DNA methylation in plants is still very rare[66].
In plant studies, it has been confirmed that changes in the DNA methylation status of multiple single genes can cause observable phenotypes, such as the Arabidopsis SUPERMAN gene[34], the CYC gene[69], the tomato CNR mature mutant[70], and the P1 gene of maize[71]. The FWA gene of A. thaliana was directly edited to show the late-flowering phenotype[66]. This gene is expected to be widely used in a variety of plant studies. Based on screening genes regulated by DNA methylation is a key biological problem of plants, systematic research on the regulation mode of genes using this technology is also the future research focus of this study.
Our future research will focus on the candidate genes of function and their promoter sequences, analysis of gene expression levels in the process of floral induction and status changes in gene promoter region methylation status changes, as high promoter region methylation and floral induction processes appear to be closely related, and we will perform targeted cytosine demethylation by CRISPR-dcas-TET1-CD.
-
The plant materials of C. lavandulifolium employed a useful strain (G1) preserved in the Chrysanthemum Breeding Laboratory in Beijing Forestry University and achieved rapid propagation under the conditions of tissue culture[72]. The G1 strain showed a quick response to short sunlight, and the morphology of the plants and capitula is shown in Fig. 5a and 5b. Collection permits from local authorities were obtained centrally by the Chrysanthemum Breeding Laboratory. C. lavandulifolium is a wild plant species native to China. These materials of C. lavandulifolium have been deposited and are publicly available in herbarium of Beijing Forestry University (deposit number BJFC00061176). When tissue culture plants attained 6−8 leaves and 5−8 roots, they were transplanted into pots. When the number of leaves of the robust plant was more than 14 (Fig. 5a), three shoots with the same growth status were taken from the stem tip of the C. lavandulifolium, the shoot tips were taken from approximately 8 leaves from top to bottom, and the sample designated the vegetative growth stage was marked as G1-0 (Fig. 5c).
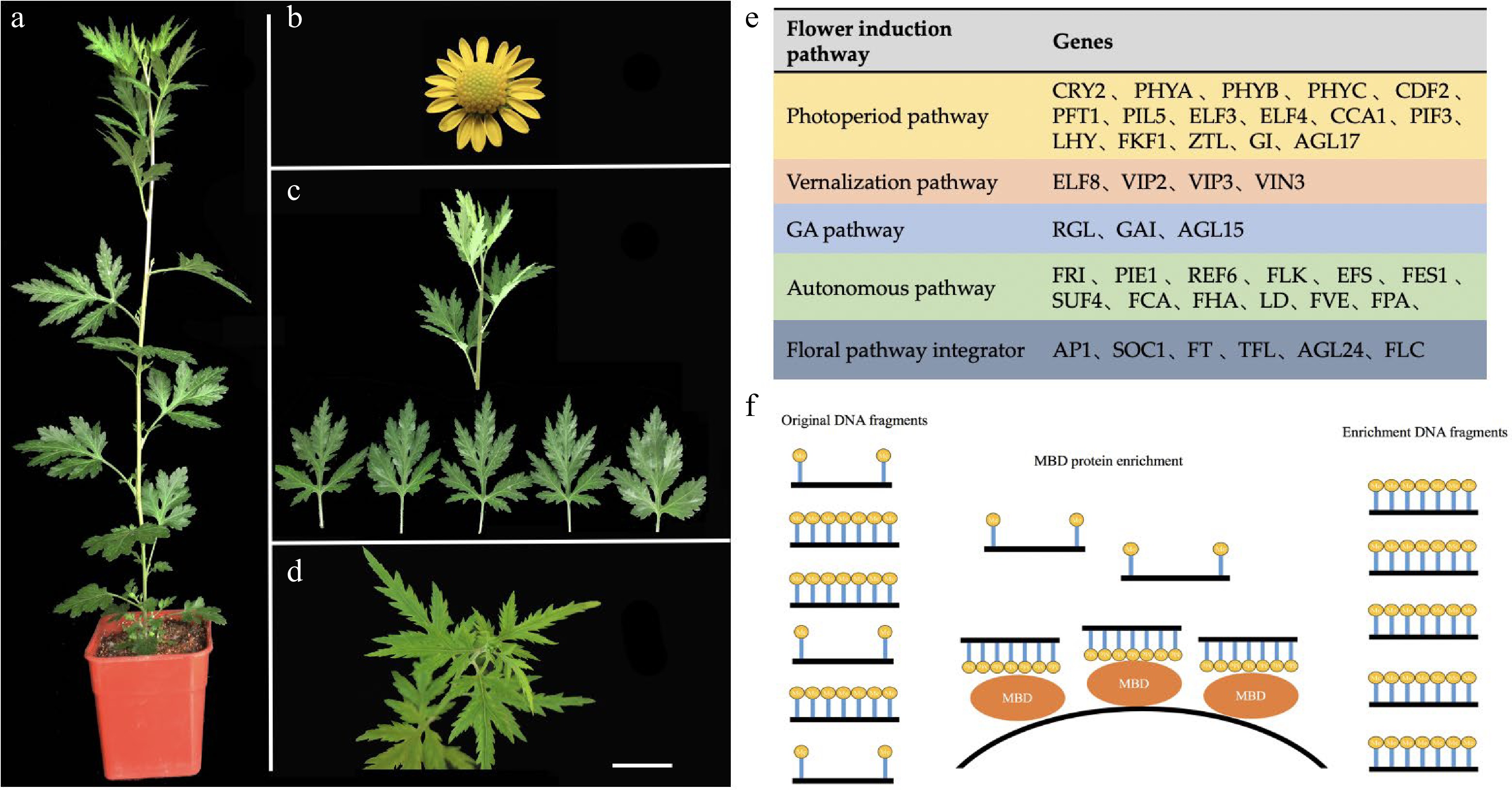
Figure 5.
Plant materials and genetic resources of Chrysanthemum lavandulifolium. (a) Mature plant with 14 leaves. (b) Capitulum. (c) Vegetative growth stage material (G1-0). (d) Bud stage material (G1-1). (e) Candidate gene resources for floral induction. (f) The model of MBD protein enrichment DNA fragment. MBD (methyl-CpG binding domain protein) can capture CpG-methylated dsDNA on beads and capture of dsDNA facilitates ligation of double-stranded adaptors for high-throughput sequencing. Bars = 1 cm.
The mature state plants (above 14 leaves) were moved to the short-day climate chamber for daily maintenance. After approximately 7 days, the plants were budded. On the 8th day, the shoot tip (approximately 8 leaves from top to bottom) was designated the bud stage and was marked G1-1 (Fig. 5d).
C. lavandulifolium produces excellent strains preserved in the laboratory and exhibits rapid propagation under tissue culture conditions with approximately 6-8 leaves and 5-8 roots when transplanting in a long-day (14 hL/10 hD) artificial climate chamber for daily maintenance.
Three strains of robust and growth-producing C. lavandulifolium with more than 14 leaves were sampled at the shoot tips (approximately 8 leaves from top to bottom) and designated the vegetative growth stage, labelled G1, and three biological replicates, marked G1-01, G1-02, and G1-03, respectively.
The mature state (above 14 leaves) was moved to the short-day (12 hL/12 hD) climate chamber for daily maintenance. After 7 days, the plants were budded. On the 8th day, the shoot tips (approximately 8 leaves from top to bottom) were named, and the bud stage was designated G1-1. Three biological replicates were taken, labelled G1-11, G1-12, and G1-13.
RT-PCR and MSP were performed on the stem tip of C. lavandulifolium (approximately 8 leaves from top to bottom) within 0−7 days of the floral induction period, and they were labelled G1-D0, G1-D1, G1-D, G1-D3, G1-D4, G1-D5, G1-D6 and G1-D7. Three biological repeats were taken for each period.
Gene sequence acquisition and CpG islands analysis
-
First, we selected the homologous gene sequence of all 42 candidate genes (Fig. 5e). These genes were derived from the flowering induction regulatory network genes of the candidate genes for the regulation of flowering date, as previously published[47]. The full length of the gene and its promoter sequence (2,000 bp upstream) were included. Sequences were obtained from the GenBank database (
www.ncbi.nlm.nih.gov ), the promoter and full-length CpG islands were analyzed.CpG islands search and site statistics were performed on all gene sequences using Methyl Primer Express Software V1.0. The CpG islands screening criteria are sequence length greater than 200 bp, C+G content greater than 50%, and CG content higher than expected 60%[3]. CG-rich regions and locations of the CpG are noted.
MBD sequencing
-
Through the Illumina HiSeq 2500 sequencing platform, the PE125 sequencing method was used to construct the MBD library of C. lavandulifolium, and then the relevant data were sequenced and obtained. The MBD protein enrichment pattern is shown in Fig. 5f. The sequencing process includes DNA extraction, DNA library construction and sequencing and analysis of DNA fragments. The sequencing process of this technology is available in detail in the MethylMinerTM Methylation DNA Enrichment Kit (Catalogue No. Me10025). The MBD sequencing process is briefly described as follows: Scale of Reactions, Elution Strategies, DNA Isolation and Fragmentation, Preparing the Beads, Incubating MBD-Beads with Fragmented DNA, Preparing Buffers for a Multi-Fraction Elution Series, Removing the Non-Captured DNA, Eluting the Captured DNA, Ethanol Precipitation, and Downstream Analysis.
Extraction of DNA and RNA and synthesis of cDNA
-
Genomic DNA was extracted using a new plant genomic DNA extraction kit (TIANGEN). The quality and concentration of DNA were detected by 1% agarose gel electrophoresis and a Nanodrop nd-2000 ultrafine assay.
Total RNA was isolated using a rapid RNA extraction kit. RNA quality and quantity were measured. RNA integrity was detected by 1% agarose gel electrophoresis. All samples were treated with RNA-free DNase I for 30 min to eliminate DNA contamination. The first cDNA strand was synthesized by an M-MLV reverse transcription system.
RT-PCR analysis
-
PCR amplification product reaction system consisted of a total of 20 comfort-l, including DNA template 1 μL, upstream primer 1 μL, downstream primer 1 μL, Easy Taq enzyme 0.3 μL, Easy Taq Buffer (10×) 2.0 μL, dNTP 0.4μL, and ddH20 14.3 μL. The reaction procedure of RT-PCR amplification was as follows: pre-denaturation at 94 °C for 5 min; denaturation at 94 °C for 50 s, optimal annealing temperature for 30 s, and extension at 72 °C for 50 s; 35 cycles were used. The internal reference gene was analyzed by ACTIN.
QRT-PCR and methylation specific PCR (MSP)
-
PCR was performed using the CFX Connect real-time System based on SYBR Premix Ex Taq. The 20-μL system included 2 μL templates (≈ 50 ng), 0.4 μL upstream and downstream primers, 10 μL 2× SYBR Premix Ex Taq and 7.2 μL ddH2O. The amplification procedure was as follows: pre-denaturation at 95 °C for 30 s; 40 cycles: denaturation at 95 °C for 5 s, optimum annealing temperature for 30 s, and extension at 72 °C for 30 s. After the completion of 40 cycles, the dissolution curve was recorded in the process from 65 °C to 95 °C, during which the temperature was maintained at 5 s for every 0.5 °C increase. All experiments were repeated three times[44].
MSP is based on bisulfite-treated DNA combined with common PCR to detect DNA methylation levels. The methylation level of CpG island in promoter sequences was detected by quantitative fluorescence PCR. Genomic DNA was extracted with plant genomic DNA extraction kit (Tiangen). The EpiTect Bisulfite Kit was used to treat DNA with bisulfite. The fluorescence quantitative PCR system of qPCR was the same as above.
Primer design
-
RT-PCR and fluorescence quantitative primers were designed using Primer software. MSP used Methyl Primer Express Software V1.0 to conduct CpG islands search and site statistics for all gene sequences and designed primers for CpG islands. Specific primer information is shown in Supplemental Table S4.
Availability of data and materials
-
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. The MBD- Seq sequencing data referred in this study were deposited in the NCBI SRA database (
www.ncbi.nlm.nih.gov/sra/PRJNA704716 ). -
During floral induction, a complex DNA methylation regulation mechanism is activated. We constructed a system of screening DNA methylation-regulated gene groups and obtained DNA methylation-regulated gene groups in the C. lavandulifolium floral induction pathway. This approach provides an effective method for related studies of epigenetics in other species without a reference genome. Based on the gene groups determined to be regulated by DNA methylation, this study supplemented the genes regulated by DNA methylation in the existing flowering regulation network (Fig. 6). Based on qRT-PCR and MSP results, it was verified that the DNA methylation changes observed on ClWRKY21 and DFL lead to their differential expression and thus regulate the flowering process of C. lavandulifolium.
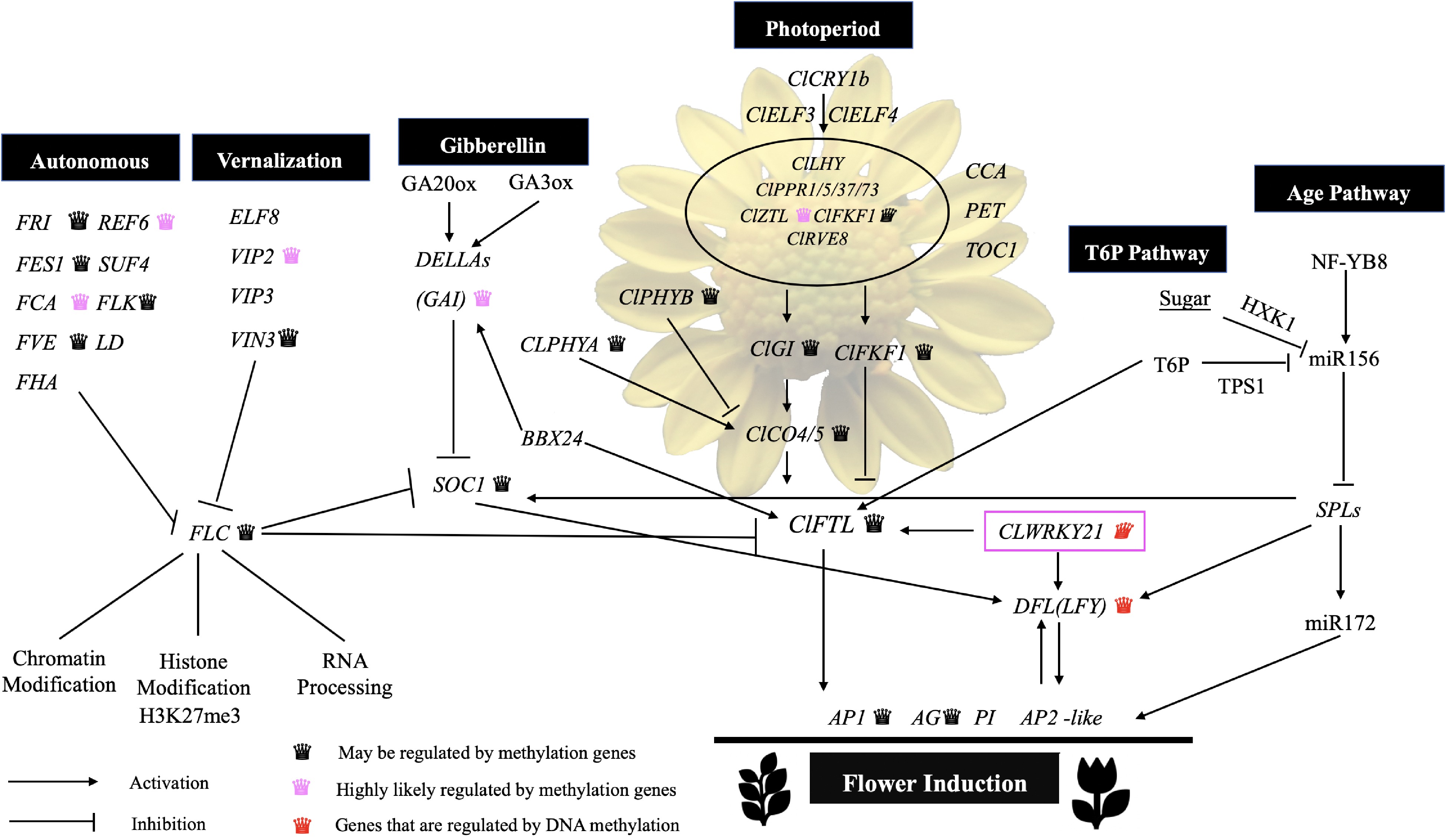
Figure 6.
Nodes in the gene regulatory network flower induction pathway may be regulated by DNA methylation.
This study was performed under the National Natural Science Foundation of China (NO. 31530064), National Key Research and Development Plan (NO.2018YFD1000403), Beijing Science and Technology Project (NO. Z191100008519002) and Major Research Achievement Cultivation Project of Beijing Forestry University (NO.2017CGP012).
-
The authors declare that they have no conflict of interest.
-
# These authors contributed equally: Dong-ru Kang, Si-lan Dai
- Supplemental Table S1 Primers for PCR analysis.
- Supplemental Table S2 CpG island screening results.
- Supplemental Table S3 Summary of promoter annotation results.
- Supplemental Table S4 Summary of mRNA annotation results.
- Copyright: © 2022 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Kang D, Dai S, Wang Z. 2022. MBD protein recognizes flower control genes regulated by DNA methylation in Chrysanthemum lavandulifolium. Ornamental Plant Research 2:3 doi: 10.48130/OPR-2022-0003 

MBD protein recognizes flower control genes regulated by DNA methylation in Chrysanthemum lavandulifolium
- Received: 30 September 2021
- Accepted: 25 January 2022
- Published online: 24 February 2022
Abstract: Dynamic changes in DNA methylation regulate the expression of genes and play important roles especially in the flowering processes of higher plants. Methyl-CpG-binding domain protein could specifically recognize hypermethylated regions in the genome, thus MBD sequencing technology and CpG islands analysis of the sequences were used to identify candidate genes that were regulated by DNA methylation, in particular the flowering induction stage of Chrysanthemum lavandulifolium. MBD-seq identified 89 candidate genes which included 49 genes exhibiting changes in DNA methylation status during floral induction. Based on CpG islands analysis of the sequences, 27 candidate genes were selected that may be regulated by DNA methylation. The expression levels of 30 candidate genes and nine key genes were determined by RT-PCR and qRT-PCR during floral induction (7D), four genes (ClFT, ClMET, DFL and ClWRKY21) were similarly up-regulated. Methylation-specific PCR analysis also indicated that there were changes in the DNA methylation status in the DFL and ClWRKY21. The changes in the DNA methylation status during the induction phase of flowering may lead to changes in gene expression. In this study, a set of genes were identified that are proposed to be involved in floral induction and two key genes were identified (DFL, ClWRKY21) that were regulated by DNA methylation during the flowering process of C. lavandulifolium.
-
Key words:
- DNA Methylation /
- MBD /
- CpG Islands /
- Floral Induction /
- Chrysanthemum lavandulifolium /
- Gene Screening