








-
Anti-counterfeiting of agricultural products plays an important role in the world market with the development of economic globalization and the rapid growth of the demand for agricultural products[1]. On the one hand, reliable anti-counterfeiting systems can increase consumers' confidence in product quality[2,3]. On the other hand, it is convenient for enterprises to supervise the quality of agricultural products and establish brands[4,5]. Because of the influence of COVID-19, consumers are more sensitive to the origin of agricultural products, and reliable anti-counterfeiting technology is urgently needed. Currently, the main food anti-counterfeiting technologies include: packaging anti-counterfeiting technology, label anti-counterfeiting technology and radio frequency identification (RFID) anti-counterfeiting technology[6,7]. These anti-counterfeiting technologies judge the authenticity by identifying the adscititious anti-counterfeiting substances (packaging, label, RF card). The disadvantage of these technologies is that consumers cannot distinguish between real anti-counterfeiting substances and imitations with an imitation degree of up to 70%, and it is difficult to prevent anti-counterfeiting substances from being used repeatedly[8,9].
Biological fingerprint refers to the behavioral and biological characteristics that can be used to identify individuals, which has been widely used in personal identification[10]. For example, human beings verify their individual identity by fingerprints, face and iris[11−13]. Agricultural products also have their own biological fingerprints[14]. With the maturity of biological fingerprint technology, it has great development prospects in the field of anti-counterfeiting of agricultural products. The biological fingerprint of agricultural products is closely related to agricultural products, which is difficult to copy and reuse. The biological fingerprint on the surface of agricultural products can be non-destructively identified by computer vision. Using biological fingerprints of agricultural products for anti-counterfeiting can effectively improve the difficulty and cost of forgery, and does not need adscititious anti-counterfeiting substances.
Lenticels are brown or white spots and stripes usually seen on the surface of plants. They are aerenchyma for plants to exchange gas with the outside world[15]. The research showed that the number and location of lenticels on mango peels were random. The location of lenticels would change due to its own size during the development of mango fruit. However, the location and size of the lenticels would hardly change after the green ripe stage[16,17]. Meaning, the features of mango lenticels are random and stable, providing good potential for biological fingerprints. However, the peel color of mango will change significantly during ripening, which may affect the recognition of mango lenticel features by computer vision.
Biological fingerprint recognition algorithms are highly dependent on the quality of the acquired biological fingerprint images[18]. Images with less obvious biological fingerprint features are usually improved using image enhancement. The enhancement process should be adaptive to the fingerprint quality to achieve the desired effect of image enhancement. Therefore, it is essential to design a quality adaptive enhancement method for biometric fingerprint images of different qualities.
Clustering refers to dividing a data set into different clusters according to a specific standard, so that the similarity of data objects in the same cluster is as large as possible, and the differences of data objects not in the same cluster are as large as possible. Among many fuzzy clustering algorithms, the fuzzy C-means clustering algorithm is the most successful and widely used[19]. It obtains the membership degree of each sample point to all class centers by optimizing the objective function, to determine the class of sample points, so as to achieve the purpose of automatic classification of sample data. Fuzzy C-means clustering algorithm is used to cluster the images with less obvious biological fingerprint features, to match the appropriate image enhancement methods to obtain better biometric fingerprint recognition effect.
-
Mango individual identity anti-counterfeiting system based on biometric fingerprints includes two parts: the establishment of identity, and the verification of identity. This workflow is shown in Fig. 1.
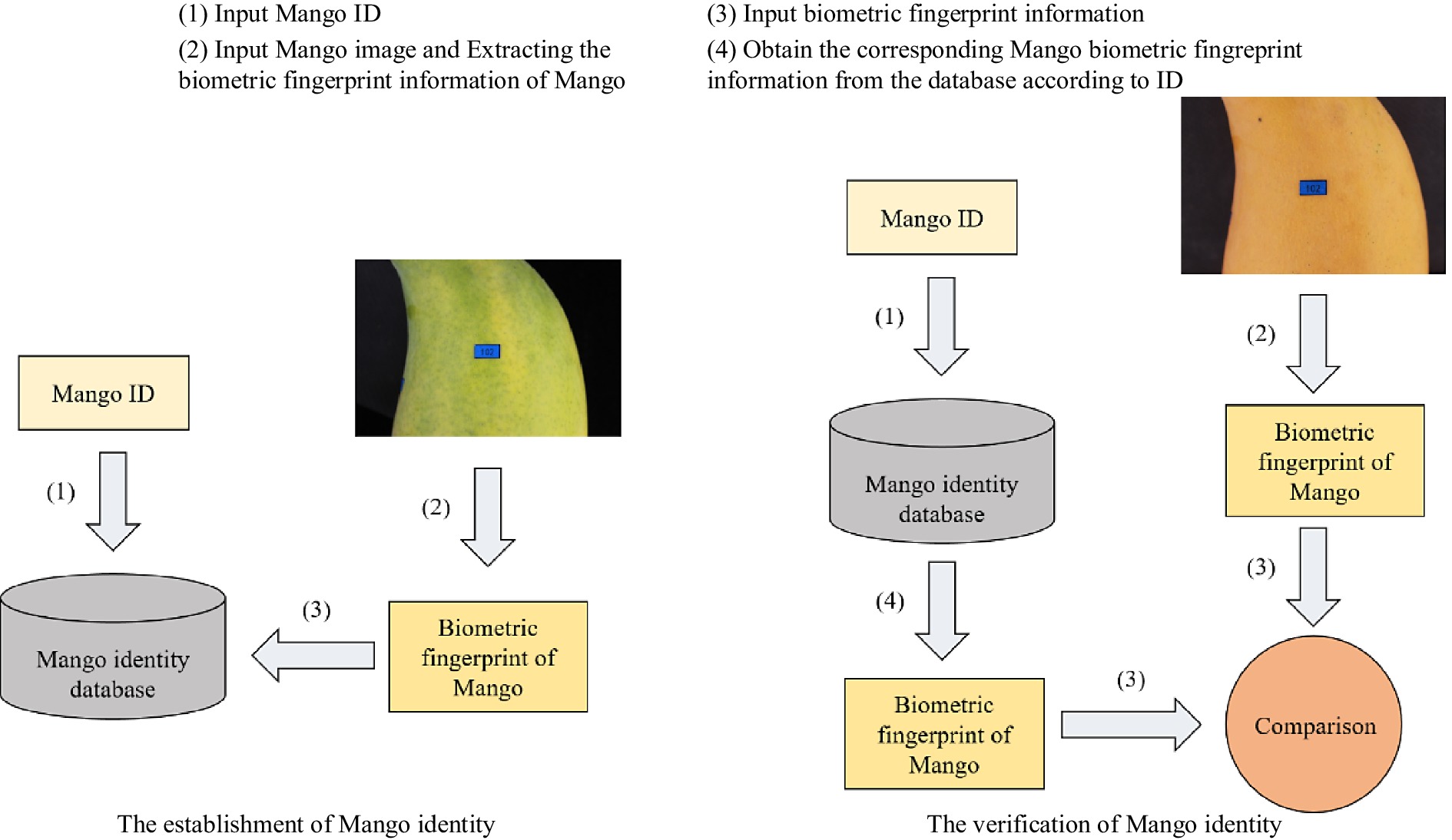
Figure 1.
Mango individual identity anti-counterfeiting system based on biometric fingerprints.
Fruit materials and treatment application
-
Two hundred and fourty green ripe mangoes (Mangifera indica L., Aroemanis) with similar maturity and no visual defects were produced in Yunnan, China, and purchased from the local market in Beijing, China. The mango sample was labeled in the center of the fruit. The serial numbers marked on the blue label were used to indicate the ID of mango individuals. These serial numbers were unique. Clear images were obtained for further experiments through the following processes:
(1) Establishment of mango identity (EI): Images of green ripe mango were acquired by camera to establish mango identity database (MIDD).
(2) Verification of green ripe mango identity (VGI): Images of green ripe mango were acquired by camera to verify the acquired images with the MIDD.
(3) Verification of ripe mango identity (VRI): After ripening mango, images of ripe mango were acquired by camera to verify the acquired images with the MIDD.
(4) Verification of ripe mango identity with different equipment (VRIE): After ripening mango, images of ripe mango were acquired by mobile phone to verify the acquired images with the MIDD.
Mangoes were stored at 25 ± 1 °C. The mango was considered as ripe when more than 3/4 of its surface were yellow.
Acquisition of images
-
Images were acquired by camera (Sony A33, 18 million pixels) and mobile phone (Huawei Mate 40, 12 million pixels;iPhone 11, 12 million pixels;Xiaomi 9, 12 million pixels) respectively. The parameters of the camera were: F/5.6, 0 exposure compensation, 1/125 s exposure time and 35 mm focal length. The main camera of mobile phone was selected. The parameters of the mobile phone were: F/5.6, 0 exposure compensation, 1/125 s exposure time and 35 mm focal length. Each mango was placed in a fixed position in the photo acquisition device. The background was set to black and illuminated by two rows of LED lights. The label was placed in the center of the image, and the four sides of the label were parallel to the four sides of the image. The images were stored on the computer in JPG format.
We obtained 230 images of EI, 230 images of VGI, 225 images of VRI and 225 images of VRIE for further studies by removing the mango with visual defects caused by storage. Then, the images were processed using MATLAB R2018a.
The time of identity establishment and identity verification is the CPU processing time using Lenovo laptop AMD core R7-4800U CPU and 16 GB RAM running Microsoft windows 10 64-bit operating system with MATLAB R2018a.
Pre-processing of acquired images
Positioning of labels
-
Extracting the blue color layer with the same color as the label could obtain the monochromatic image with obvious features of the label. Since only the label in the image was blue, the binary image of the label could be obtained by binarizing the monochrome image at a certain threshold. The binary image was optimized by opening operation to remove the noise, so that only the relevant features of the label were retained. Thus, the position and size of the label area were obtained.
Image normalization
-
After the label was positioned, the original image (Fig. 2a) was rotated and scaled according to the position and size of the label, ensuring the four sides of the label were parallel to the four sides of the image. The label size was unified to 60 × 100 pixels. Then through cutting processing, we finally obtained the scale normalized image (220 × 220 pixels, Fig. 2b) with the label (60 × 100 pixels) in the center.

Figure 2.
Image processing. (a) Original image. (b) Scale normalized image. (c) Gray normalized image. (d) Binary image.
Although we reduced the change of illumination as much as possible, the image was still affected by the illumination angle and other factors. The gray value and variance of the image needed to be normalized to a specific range after the image was transformed into a gray image, so as to reduce the influence of the gray level change caused by the change of illumination on the identification effect[20,21]. The image was divided into 100 blocks (26 × 26 pixels). The gray average and variance of each block were calculated and expressed in MX and VX. M0 and V0 were the expected gray average and variance. The I(i, j) was the gray value of the corresponding coordinate pixel in the image. The G(i, j) was the normalized gray value. The equations were as follows:
$ G\left(i,j\right)={M}_{0}+\frac{\sqrt{{{V}_{0}[I\left(i,j\right)-{M}_{x}]}^{2}}}{{V}_{x}} \quad I (i, j) \gt Mx $ (1) $ G\left(i,j\right)={M}_{0}-\frac{\sqrt{{{V}_{0}[I\left(i,j\right)-{M}_{x}]}^{2}}}{{V}_{x}} \quad I (i, j)\leq Mx $ (2) To smooth noise and preserve the edges of the images, median filtering was adopted[22]. Finally, the normalized image was obtained (Fig. 2c).
Feature extraction
-
Owing to the different colors of mango lenticels and a lot of redundant information in the background area, if the whole image is binarized with the same threshold, a lot of useful information will be lost. For better feature segmentation, the image was binarized using the adaptive threshold method[23,24]. The binary image was optimized by opening operation to remove the noise or instability features left in the image. In the end, we would gain clear binary images (220 × 220 pixels, Fig. 2d). Record the coordinates of the feature area in the binary image, which contains the information of the position and size of mango lenticels, that is, the mango lenticel feature.
Because the peel color of mango changed after ripening, we modified several parameters in image preprocessing and feature extraction for mango in different ripening states, and other methods were completely consistent.
Feature contrast and similarity value calculation
-
The image of EI was used as the standard image. The position and size of lenticels were used to establish the MIDD. After the mango image which needed to be verified were processed as described above, the obtained lenticel feature could be compared with the lenticel feature of the corresponding identity mango in the MIDD. If two lenticels from different images partially coincided, the matching of the lenticel was regarded as successful.
The similarity value was calculated by the successfully matched lenticel area divided by the sum of the lenticel area of the two images.
Fuzzy C-means clustering
-
One hundred and seventyfive images of VRI and 175 images of VRIE were randomly selected for Fuzzy C-means clustering. The remaining 50 images of VRI and 50 images of VRIE were tested for identification. The images used for Fuzzy C-means clustering would be preprocessed to obtain the normalized image. Then, the following operations were carried out.
Cluster characteristic selection
-
We selected some relevant characteristics, which can define the image quality to cluster the normalized image into different quality levels.
(1) Average value: the average value of the gray value of all pixels in the image.
(2) Variance: the variance of the gray value of all pixels in the image.
(3) Maximum gray value: the maximum gray value of all pixels in the image.
(4) Minimum gray value: the minimum gray value of all pixels in the image.
(5) Contrast: contrast refered to the ratio between the brightest white and the darkest black in the image, that is, the gradient level of the grayscale of the image. A large contrast value represented more gradient levels and richer color expression.
$ C=\mathop\sum\nolimits _{\delta }\delta (i,j{)}^{2}{P}_{\delta }(i,j) $ (3) Where,
$ \delta (i,j) $ $ {P}_{\delta }(i,j) $ $ \delta (i,j) $ (6) Uniformity: the uniformity of the distribution of image pixels on the gray histogram.
$ U=1-\dfrac{\displaystyle\mathop\sum\nolimits _{i=0}^{255}\left|{r}_{i}-s/255\right|}{\displaystyle\mathop\sum\nolimits _{i=0}^{255}\left({r}_{i}+s/255\right)} $ (4) Where, ri represented the number of pixels in the image with gray value i, and s represented the sum of gray values of all pixels in the image.
(7) One-dimensional entropy: the image entropy reflected the average amount of information in the image. The one-dimensional entropy of an image represents the amount of information contained in the aggregated characteristics of the gray distribution of the image.
$ H=-\mathop\sum\nolimits_{i=0}^{255}{P}_{i}\mathit{log}{P}_{i} $ (5) Where, Pi represented the proportion of pixels in the image whose gray value was i.
(8) Two-dimensional entropy: in order to characterize the spatial characteristics of the gray distribution of the image, one-dimensional entropy and the characteristic quantity that could reflect the spatial characteristics of the gray distribution form the two-dimensional entropy of the image.
$ H=-\mathop\sum\nolimits _{i}\mathop\sum\nolimits _{j}{P}_{i,j}\mathit{log}{P}_{i,j} $ (6) Where, Pi,j represented the proportion of pixels with gray value was i and domain mean value was j in the image.
The above characteristics needed further screening. Each cluster characteristics of 350 normalized images were calculated respectively, the cluster characteristics were then normalized. The characteristics with low coefficient of variation (less than 0.15) were removed and then the similarity between the remaining characteristics were calculated, respectively. If the two characteristics were significantly correlated (the absolute value is greater than 0.6), only one characteristic would be retained. Finally, three cluster characteristics were obtained which could be used to classify image quality including the variance, the minimum gray value, and the contrast.
Fuzzy C-means clustering
-
According to the clustering characteristics obtained in the above steps, 350 normalized images were clustered. They were divided into three categories (I, II and III) by using the Fuzzy C-means clustering algorithm. The three clustering centers were recorded. When the image to be verified needed quality classification, the membership degree between it and the three clustering centers could be calculated to judge the quality category it belonged to[25].
Evaluation of the performance of the identification method
-
The method proposed in this paper was to calculate the lenticel feature similarity between the mango to be verified and the mango with the same ID in the database, judging the authenticity of mango identity. When the VGI, VRI and VRIE groups were verifying the identity of mango, we input the correct mango ID as the positive sample and the wrong mango ID as the negative sample. The performance of the mango identification method was evaluated by the Receiver Operating Characteristic (ROC) curve, the Equal Error Rate (EER) and the verification time (the total time of image reading, image preprocessing, feature extraction and similarity calculation). The ROC curve was a graph of the True Positive Rate (TPR) and the False Positive Rate (FPR). The TPR and FPR were defined as following:
$ \text{TPR = }\frac{\text{TP}}{\text{TP + FN}} $ (7) $ \text{}\text{FPR = }\frac{\text{FP}}{\text{FP + TN}} $ (8) The definition of TP, FP, TN and FN are shown in Table 1.
Table 1. Prediction results of the identification method.
Positive sample Negative sample Correct identification TP (True positive) FP (False positive) Error identification FN (False negative) TN (True negative) The EER was the point on the ROC curve corresponding to an equal probability of error identification for a positive or negative sample. This point was obtained by intersecting the ROC curve with a diagonal of the unit square. In other words, the EER was the FPR value when TPR + FPR = 1.
-
We only saved the lenticel feature information of mango image in the database. On average, the feature information of each mango only needed to occupy about 360 bytes of memory. Assuming that each mango weighs 0.25 kg, 1000 tons of mango only needed 1.4 GB of memory to back up and archive its identity information. In addition, the time of identity establishment (the total time of reading images, image preprocessing, feature extraction and writing to the database) only took about 0.60 s per mango. Therefore, this method could store and identify the identification information of mango with huge yield. Moreover, this method did not need a lot of image training. Compared to image identification by deep learning, the method reduced the computing power consumption and memory consumption significantly[26,27].
Conventional mango biological fingerprint anti-counterfeiting method
-
We performed image preprocessing and feature extraction for 230 VGI, 225 VRI and 225 VRIE respectively. The extracted features were compared with the features of 230 EI in MIDD. The results were as follows:
According to Fig. 3 and Table 2, the area under curve (AUC) value of the ROC curve obtained by the VGI group was very close to 1, and the EER was very close to 0%, indicating that the lenticel distribution on the mango fruit surface were unique. The changes in the shooting angle and shooting height within a certain range had little impact on the identification effect. Compared to the results of the VRI and VGI group, we could find that the change of the peel color of the mango during the ripening process did impact the identification rate greatly of this method. However, the AUC value above 0.98 and the EER within 5% indicated that the mango identity could be identified by this method as long as the fruit did not have obvious visual defects, proving that the lenticels distribution on the surface of mango fruit had good stability. In the VRIE group, we used three different mobile phones to acquire images. Through comparison with the VRI group, we found that different shooting equipment from the EI group would reduce the accuracy of identification.
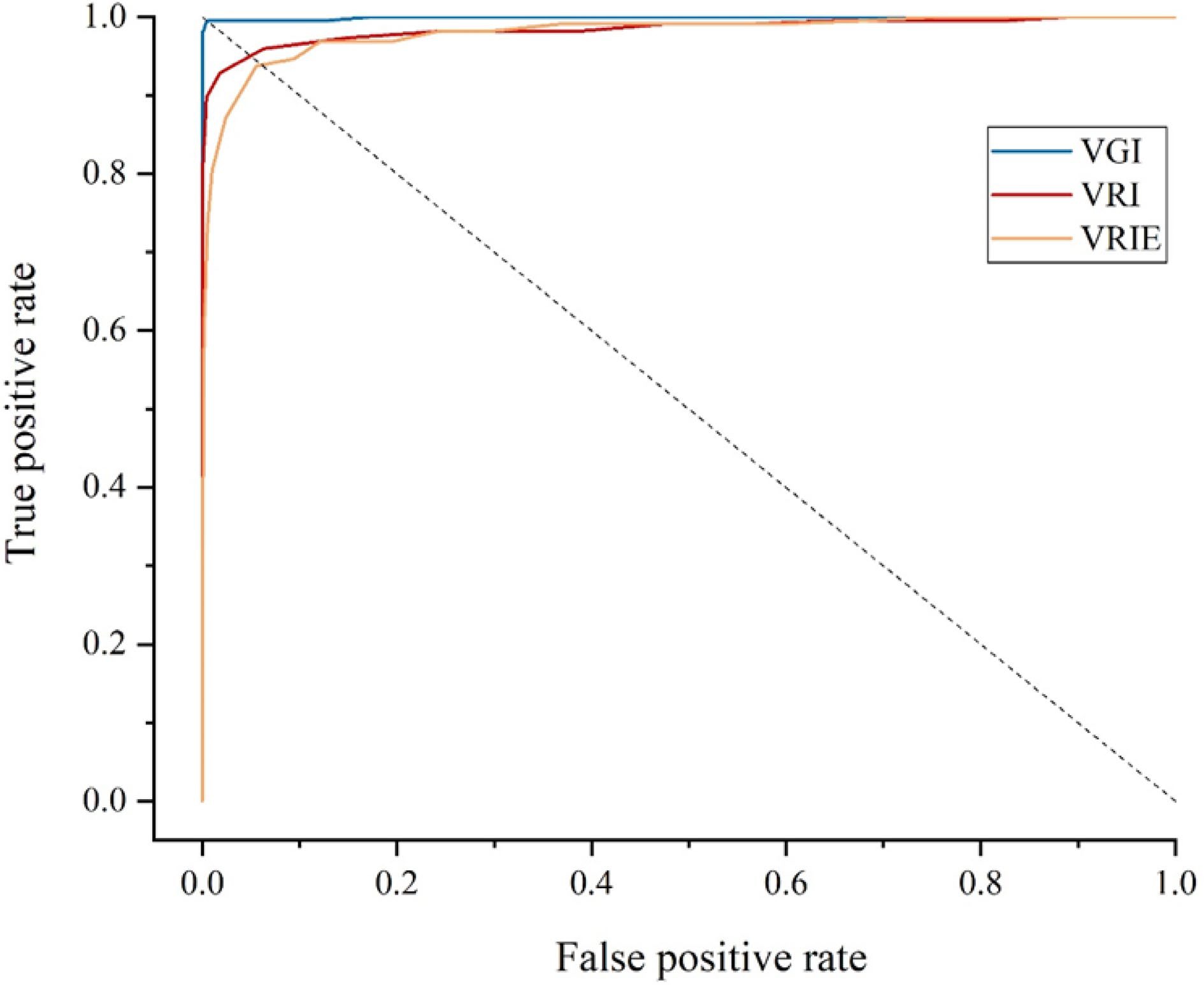
Figure 3.
The ROC curves of conventional mango identification methods.
Table 2. Performance of conventional mango identification methods.
Verification time (s) AUC EER (%) VGI 0.6195 0.9993 0.43 VRI 0.6058 0.9853 4.80 VRIE 0.3993 0.9805 6.23 The average verification time of the three groups of images are shown in Table 2. According to the above identification method, the verification time of the images taken by the camera was about 0.61 s. The difference in pixel size of the original image captured by different photographing equipment affected the verification time significantly. Larger original images required longer verification time.
The above results proved the feasibility of anti-counterfeiting via the lenticel features of mango as a biological fingerprint. However, we also observed that the accuracy and robustness of this method need to be improved. The identification method needed to be further optimized to improve the effect.
Mango biological fingerprint anti-counterfeiting method based on Fuzzy C-means clustering
-
In order to further improve the accuracy of the anti-counterfeiting method and reduce the influence of peel color changes during mango ripening, the image enhancement technology was used to highlight features to make it easier to extract and identify. However, due to the differences in image quality and ripening process of each mango individual, a single image enhancement technology could not meet the needs. Therefore, we used Fuzzy C-means clustering to classify the images to be verified into three categories according to the image quality. After the image was pre-processed, we calculated its membership degree with the three clustering centers to judge its quality category. Then, the corresponding image enhancement technology was carried out to facilitate the subsequent feature extraction.
One hundred and seventyfive images of VRI and 175 images of VRIE were randomly selected for Fuzzy C-means clustering. The remaining 50 images of VRI and 50 images of VRIE were identified. The obtained results were expressed as VRIC and VRIEC.
We clustered 350 normalized images into three categories through fuzzy C-means clustering. These three categories of images were different from the minimum gray value, variance and contrast (Fig. 4). After Fuzzy C-means clustering, we found that the features of class I images were relatively obvious, and the feature extraction could be carried out well without image enhancement. The features of class II images were similar to the background gray values, which were difficult to extract. According to Fig. 4, we found that the minimum gray value and contrast of class II images were significantly smaller than those of class I images. We could use histogram equalization algorithms to improve the gap between feature and background[28]. The background part of class III images had more redundant information, which affected feature extraction. According to Fig. 4, we found that the gray variance and contrast of class III images were significantly larger than those of class I images. We use wavelet transform for image enhancement. After the original image was decomposed into images with different frequencies, we used various methods to enhance images with different frequencies, so that reducing the influence of the background part to improve this situation[24,25].
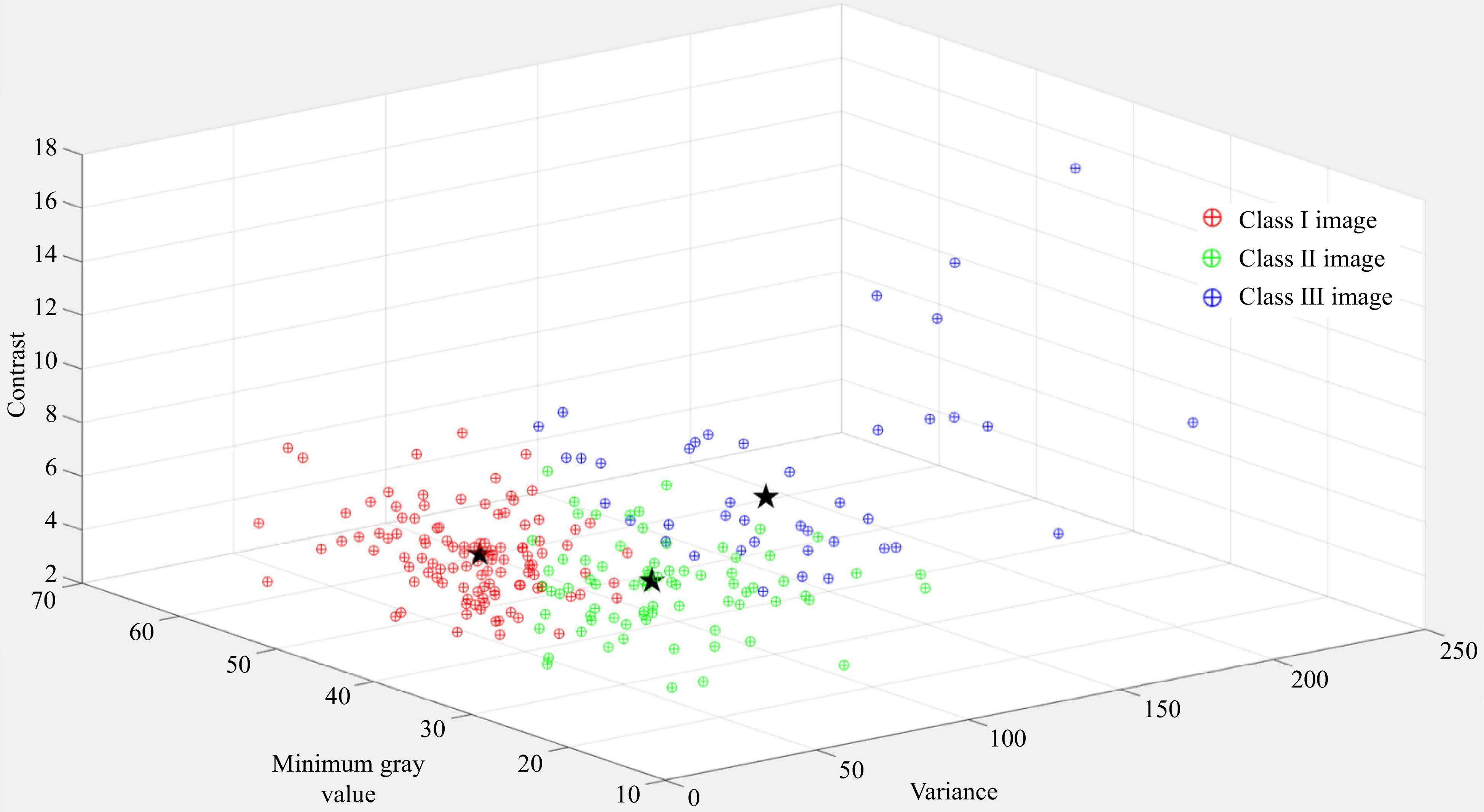
Figure 4.
Fuzzy C-means clustering and clustering center of 350 normalized images.
According to Fig. 4, the Fuzzy C-means clustering algorithm successfully clustered the images into three categories. According to Fig. 5 and Table 3, it can be seen that the impact on the changes of mango peel color could be reduced by using appropriate image enhancement techniques for different categories of images in the biometric fingerprint identification method. These strategies improved the accuracy and robustness of the method. After the image was enhanced, the AUC value of VRIC group was significantly improved and the EER was significantly decreased, which proved that the change of mango peel color had limited impact on the biometric fingerprint identification method. And the application scope of biometric fingerprint anti-counterfeiting method was broadened through our strategies. Compared to VRIE group, VRIEC group also reduced the impact significantly of different shooting equipment on accuracy. The presented results showed that the method had low equipment requirements. The consumers and administrators could simply complete image acquisition through a smart phone. The presented method of Fuzzy C-means clustering was very convenient for anti-counterfeiting.
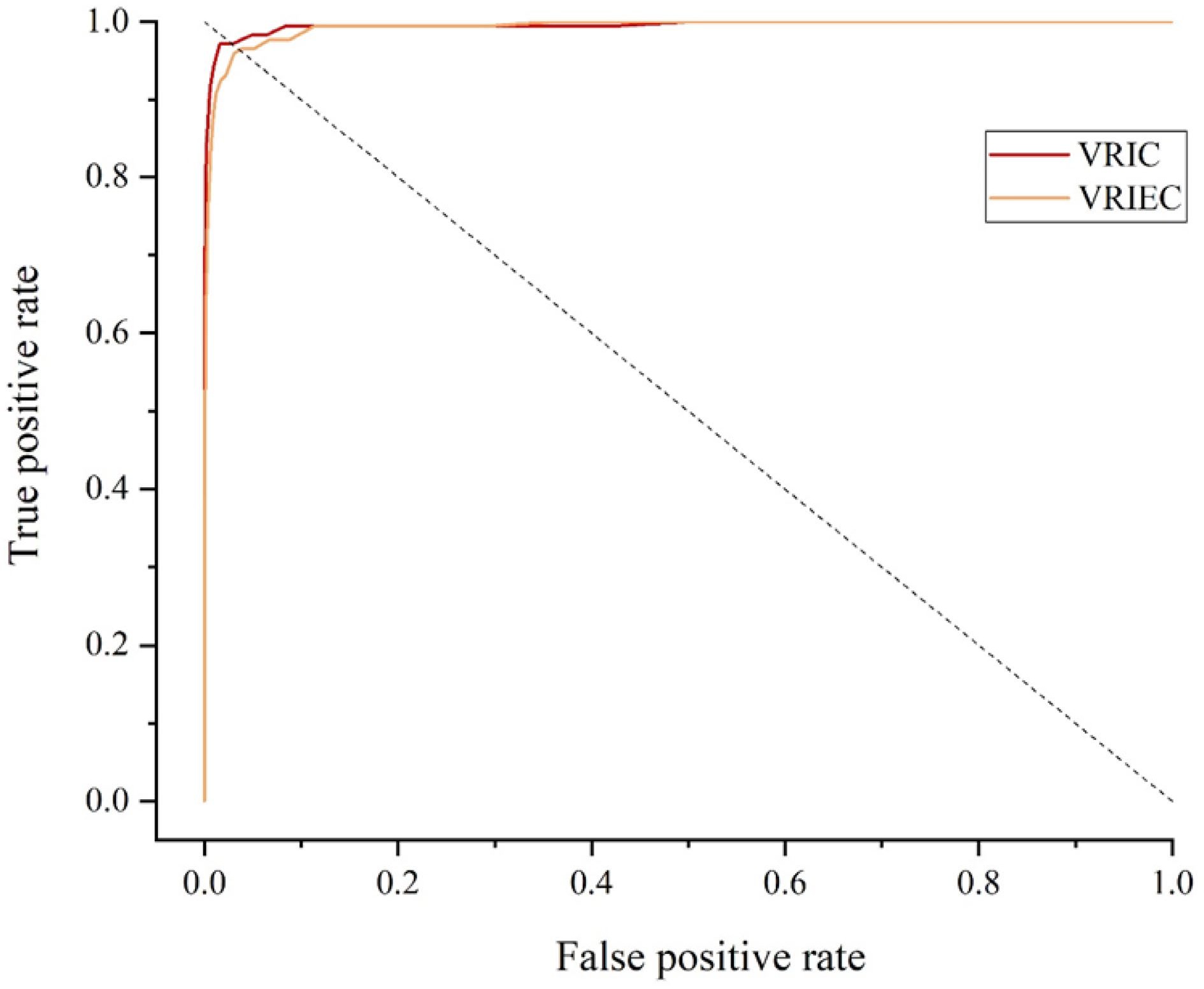
Figure 5.
ROC curves of the mango identification method based on Fuzzy C-means clustering.
Table 3. Performance of the mango identification method based on Fuzzy C-means clustering.
Verification time (s) AUC EER (%) VRIC 0.6326 0.9950 2.86 VRIEC 0.4212 0.9930 3.43 -
This research proved the feasibility of taking the feature of mango lenticels as biological fingerprints. The changes of mango peel color had a limited influence on biological fingerprint identification methods. We effectively reduced the impact of the change of mango peel color on the biometric fingerprint identification method by the Fuzzy C-means clustering algorithm. A computer vision anti-counterfeiting method was proposed based on mango lenticel features, revealing the potential of the biometric fingerprint anti-counterfeiting recognition by using the surface features of agricultural products.
This work was supported by the National Natural Science Foundation of China (No. 32172270).
-
The authors declare that they have no conflict of interest.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press on behalf of China Agricultural University, Zhejiang University and Shenyang Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Shen C, Zhang Y, Chen L, Jia A, Cao J, et al. 2023. A mango biological fingerprint anti-counterfeiting method based on Fuzzy C-means clustering. Food Innovation and Advances 2(1):21−27 doi: 10.48130/FIA-2023-0004 

A mango biological fingerprint anti-counterfeiting method based on Fuzzy C-means clustering
- Received: 08 September 2022
- Accepted: 05 December 2022
- Published online: 20 February 2023
Abstract: The anti-counterfeiting of agricultural products plays an important role in protecting the rights and interests of consumers and maintaining the healthy development of the food market. Traditional anti-counterfeiting technology mainly relies on anti-counterfeiting features of packaging or labeling, which has the risk of being copied and reused. Biological fingerprint anti-counterfeiting is a method of anti-counterfeiting that takes the biological fingerprint of agricultural products as the anti-counterfeiting feature. This paper aims to take the distribution of lenticels on the surface of mango as a biological fingerprint, and propose a mango biological fingerprint anti-counterfeiting method. As the mango ripens, the peel color of mango will change significantly, which will affect the accuracy of anti-counterfeiting identification. In this paper, the images of ripe mangoes are classified by Fuzzy C-means clustering, and appropriate image enhancement technology is used to highlight the features. The results show that the mango biological fingerprint anti-counterfeiting method based on Fuzzy C-means clustering has good accuracy and robustness, and effectively reduces the impact of peel color change on anti-counterfeiting identification during mango ripening. These results support that it is feasible to use the lenticels distribution of mango as a biological fingerprint. In this paper, a computer vision anti-counterfeiting method based on lenticels distribution is proposed.