









-
Kombucha, a widely consumed beverage, is celebrated for its abundant probiotics and bioactive compounds. This fermented drink is traditionally prepared by fermenting tea leaves and sucrose with a symbiotic culture of bacteria and yeast (SCOBY)[1]. The unique blend of microorganisms and chemical constituents in kombucha is fundamental to its reported health advantages, which range from improving gastrointestinal health to exhibiting anti-inflammatory, antioxidant, anticancer, cardiovascular protective, and therapeutic properties in conditions such as rheumatism, gout, and hemorrhoids[2−6].
The fermentation process of kombucha is a symbiotic relationship where yeast converts the sugar into monosaccharides like glucose, primarily to produce ethanol, while acetic acid bacteria subsequently transform this ethanol into acetic acid[7,8]. As fermentation unfolds, the flavor and health-promoting compounds in kombucha continue to develop, yielding a complex matrix of small molecular metabolites. The intricate and diverse nature of the fermentation process has sparked considerable interest in the microbial community of kombucha, its preparation methods, and its utility across different sectors.
While black tea is the traditional base for kombucha, other varieties such as green tea, Oolong tea, and dark tea are also employed. Recently, there has been a surge in interest in using Pu-erh tea or Fuzhuan brick dark tea to produce dark tea kombucha[9−11]. However, the scientific literature on the microbial composition and chemical constituents of dark tea kombucha remains sparse. Existing research has predominantly focused on the antioxidant potential and polyphenol content of kombucha[12−14]. In this context, the use of Liupao tea was chosen for investigation, a distinguished dark tea, as a base for kombucha production. Known for its probable prebiotic effects and esteemed as a superior fermented dark tea, Liupao tea undergoes a unique post-fermentation process that not only amplifies its microbial diversity but also intensifies its polyphenolic composition[15]. It is renowned for a spectrum of health benefits, including gut microbiota improvement, antioxidant activity, and glucose and lipid metabolism regulation[16−18]. These attributes are anticipated to endow the kombucha with a richer and potentially healthier profile, suggesting Liupao tea is an ideal and promising substrate for dark tea kombucha fermentation.
This study was designed to prepare three distinct types of kombucha using various substrates, including Liupao tea from two different vintages, which are novel substrates, alongside conventional green tea. Sensory evaluations, microbial sequencing, and widely targeted metabolomics were conducted to examine the microbial diversity and key differential metabolites in the kombucha fermented with Liupao tea (K-7LP). The objective was to provide a comprehensive assessment of the microbial composition and metabolite profile of dark tea kombucha. Furthermore, network pharmacology approaches were utilized to dissect the pivotal bioactive components in Liupao tea kombucha. Through these extensive analyses, the aim is to to broaden and deepen the understanding of dark tea kombucha, illuminating its potential benefits and applications.
-
Green tea was sourced from the local market. Dark tea, specifically Liupao tea aged for 1 year and 7 years, was obtained from Wuzhou Tea Factory in Guangxi, China. The Kombucha SCOBY (Symbiotic Culture of Bacteria and Yeast) was purchased from Beijing Sisi Slow Food Catering Co., Ltd (Beijing, China). The main equipment used included an incubator (LBI-250).
Preparation and cultivation of kombucha
-
The procedure for preparing kombucha in this study involved the following specific steps: First, 5 g each of green tea, one-year-aged Liupao tea, and seven-year-aged Liupao tea were separately weighed. The tea samples were then steeped in 500 mL of boiling water for 3 min, followed by filtration to remove the residues, yielding the tea infusions. These tea infusions were transferred to sterile 1,500 mL glass fermentation vessels, and 50 g of sugar was added to each and stirred until fully dissolved. After the infusions cooled, 20 g of kombucha SCOBY and 50 mL of kombucha stock solution were added. The vessels were then covered with clean, triple-layered cheesecloth and placed in a biochemical incubator set to a temperature of 28 °C and a humidity of 30%−40%. Fermentation was carried out for 14 d, resulting in green tea kombucha (K-GT), one-year-aged Liupao tea kombucha (K-1LP), and seven-year-aged Liupao tea kombucha (K-7LP). Finally, 30 mL samples of K-GT, K-1LP, and K-7LP were flash-frozen in liquid nitrogen for 5 min and stored at −80 °C for subsequent analysis.
16S and ITS bioinformatics analysis
-
The kombucha samples K-GT, K-1LP, and K-7LP were analyzed by BGI Genomics Company. The PCR reaction system was configured with 30 ng qualified genomic DNA sample and corresponding fusion primers, the corresponding PCR reaction parameters were set for PCR amplification, and the PCR amplification products were purified to complete the construction of the library. The fragment range and concentration of the library were measured using Agilent 2100 Bioanalyzer. Qualified libraries were sequenced on BTU's autonomous DNBSEQ platform. The downstream data were filtered to remove low-quality reads, and the remaining high-quality clean data could be used for later analysis; the reads were spliced into Tags through the Overlap relationship between the reads; the Tags were clustered into OTUs under the given similarity, and then the OTUs were compared with the database to perform the species annotation of the OTUs; the species complexity of the samples was analyzed based on the results of OTUs and species annotation; and the species annotation of the samples was performed based on the results of OTUs and species annotations. Species annotation results were used to analyze the species complexity of the samples and the species differences between groups.
Microbial diversity was assessed using the sobs index to provide a comprehensive view of microbial richness and evenness in the kombucha samples. The Kruskal test was applied to identify specific pairwise differences. LEfSe (LDA Effect Size) analysis was conducted using the LEfSe tool (
https://huttenhower.sph.harvard.edu/galaxy/ ), to identify statistically significant differences in microbial populations among the samples. In this study, the parameters used in LEfSe analysis included: α value of 0.05, which was used to identify microbial communities with statistically significant differences; The LDA threshold is 2.0, which is used to screen biomarkers that have a significant impact on classification. The non-parametric factorial Kruskal-Wallis (KW) sum-rank test was used to detect features with significant differential abundance with respect to the class of interest; biological significance was subsequently investigated using a set of pairwise tests among subclasses using the (unpaired) Wilcoxon rank-sum test. As a last step, LEfSe uses Linear Discriminant Analysis to estimate the effect size of each differentially abundant feature.Widely targeted metabolomics analysis
-
The K-GT and K-7LP samples were analyzed using widely targeted metabolomics with Ultra-High Performance Liquid Chromatography-Tandem Mass Spectrometry (UHPLC-MS/MS). This widely targeted metabolome analysis was conducted at Metware Biotechnology Co., Ltd. in Wuhan, China, to identify the metabolic differences between K-GT and K-7LP. The samples were thawed on ice after being removed from the −80 °C freezer. After vortexing for 10 s, 200 μL of the sample was collected and centrifuged at 4 °C for 10 min. The supernatant was filtered through a 0.22 μm microfiltration membrane and stored in the injection vial. The UPLC-MS/MS instrument system was used for data acquisition, consisting of an ultra-performance liquid chromatography system and a tandem mass spectrometry system. The liquid phase conditions were as follows: chromatographic column: Agilent SB-C18 1.8 μm, 2.1 mm × 100 mm; mobile phase: A (ultrapure water with 0.1% formic acid) and B (acetonitrile with 0.1% formic acid); gradient elution: B proportion increased from 5% to 95% within 9.00 min, maintained at 95% for 1 min, decreased to 5% from 10.00 to 11.10 min, and equilibrated at 5% until 14 min; flow rate: 0.35 mL/min; column temperature: 40 °C; injection volume: 2 μL. The mass spectrometry conditions were as follows: electrospray ionization source temperature: 500 °C; ion spray voltage (IS): 5,500 V (positive ion mode)/−4,500 V (negative ion mode); ion source gas I (GSI), gas II (GSII), and curtain gas (CUR) were set at 50, 60, and 25 psi, respectively; collision-induced dissociation parameters were set to high.
The raw data were processed using the Analyst 1.6.3 software (AB Sciex, Framingham, MA, USA). Metabolomics multivariate statistical analysis was performed using SIMCA-P 14.1 software (Umetrics Corporation, Umea, Sweden). Variable importance in projection (VIP) values for all metabolites from the OPLS-DA were extracted from the first component. Differential metabolites between KGT and KLP were identified based on a fold change of ≥ 2 or ≤ 0.5, with a significance level of p ≤ 0.05. Additionally, Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation and metabolic pathway analysis were performed for these metabolites. A hypergeometric test was employed to identify significantly enriched pathways (p < 0.05).
Correlation analysis between microorganisms and metabolites
-
The correlation analysis between microorganisms and metabolites was conducted using OmicStudio (
www.omicshare.com/tools , accessed on 17 Feb 2024). The mmvec method was employed using the QIIME2 pipeline to analyze the correlations between microbial abundances and specific metabolites. The Z-score (zero-mean normalization; z = (x − μ)/σ) was employed, where x is the original value, μ is the mean, and σ is the standard deviation) of the principal differential metabolites and key microorganisms enriched in the top five KEGG pathways of two kombucha variants to assess their correlation. The data was first tested for normality. The Shapiro-Wilk test was used to evaluate the normal distribution of the data set. The results showed that all the datasets participating in Pearson correlation analysis had p-values greater than 0.05. A heat map was then generated using Pearson correlation coefficients with the correlation heat map tools available in OmicStudio.Network pharmacological analysis
-
Based on the differential metabolites provided by the metabolomics test results (VIP ≥ 1, fold change ≥ 2 or ≤ 0.5), the compounds were converted into Simplified Molecular Input Line Entry System (SMILES) through the PubChem database (
https://pubchem.ncbi.nlm.nih.gov/ ). The selected compounds were input into SWISSADEM (www.swissadme.ch ). Compounds with a 'Bioavailability Score > 0.17' were selected as active compounds with good bioavailability. The targets of the compounds were predicted through SwissTargetPrediction (www.swisstargetprediction.ch ), with a threshold setting of Probability > 0 as the screening criterion. The genes were imported into STRING (https://string-db.org ), with a Combined score ≥ 0.9 as the screening standard, to obtain predicted Protein-Protein Interactions (PPI). Then, genes with a Degree value greater than the average node degree were visualized and a key active component-target network map was established using Cytoscape 3.9.1 (https://cytoscape.org/ ). These target genes were further analyzed using online functional annotation and enrichment pathway analysis in Metascape (www.metascape.org ), selecting Homo sapiens as the species. Intersection targets underwent KEGG pathway enrichment analysis, and the top 20 results were visualized. -
The species accumulation curves (Fig. 1a) indicate that the sampling size for this experiment was adequate, making the data suitable for further analyses. Figure 1b illustrates the overlap of Operational Taxonomic Units (OTUs) across the three sample groups. Among these groups, 30 bacterial OTUs were shared, with K-7LP having 152 unique OTUs, K-1LP with 49 unique OTUs, and K-GT with six unique OTUs. The results suggest that K-1LP and K-7LP exhibit higher bacterial diversity compared to K-GT, as shown in Fig. 1c.
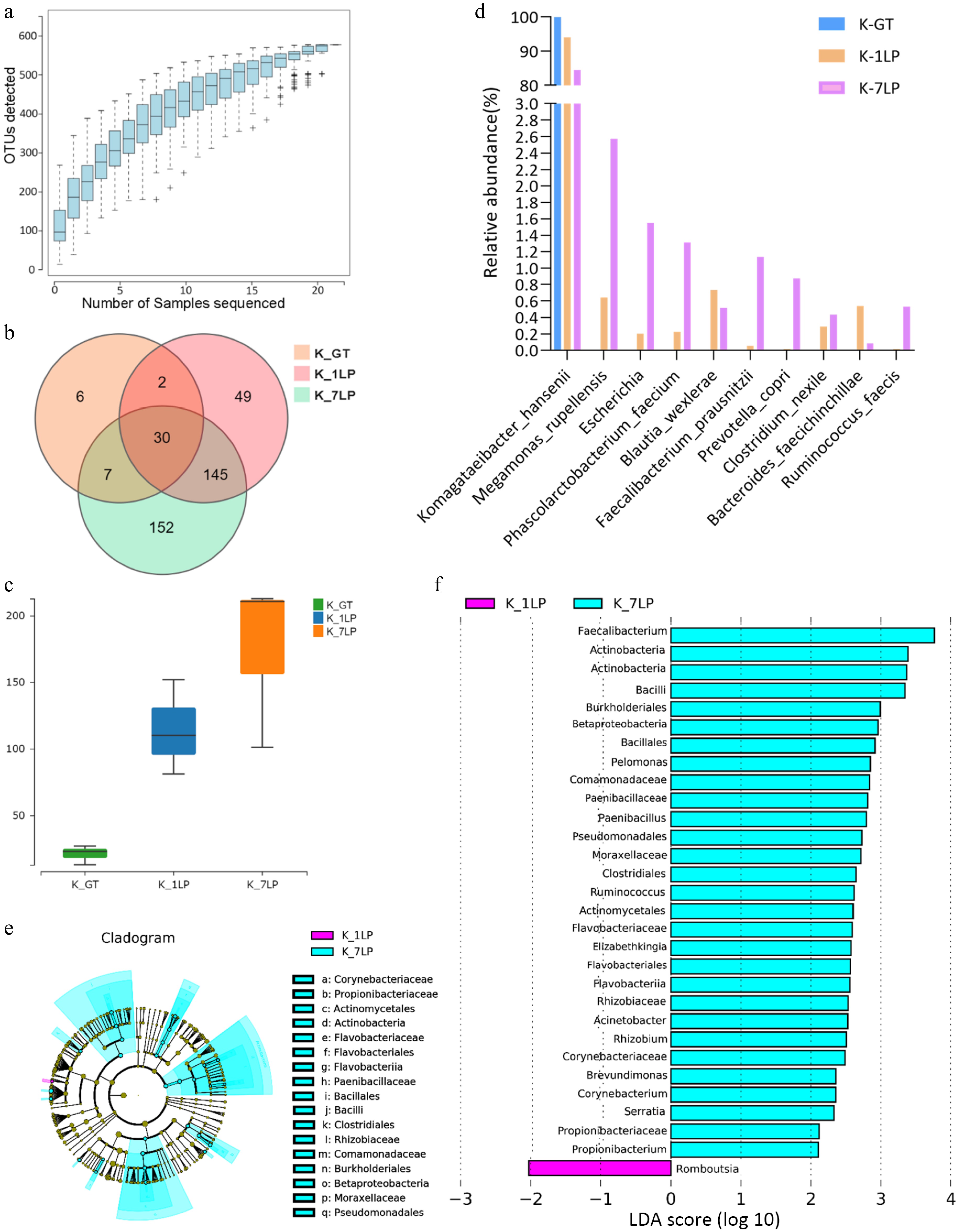
Figure 1.
Statistical analysis of 16S sequencing data for three kinds of kombucha. (a) Species rarefaction curve; (b) Venn diagram of bacterial OTUs; (c) Boxplot of α-diversity (sobs index). The Kruskal Test test is used to show differences in alpha diversity between groups. The five lines from bottom to top are: minimum, first quartile, median, third quartile, and maximum; (d) Histogram of relative abundance of three kinds of kombucha key bacteria; (e) Evolutionary branch diagram. Different colors indicate different groups, and nodes in different colors indicate microbiomes that play an important role in the group represented by the color. A color circle represents a biomarker, and the legend in the upper right corner is the biomarker name. The yellow nodes represent microbial groups that do not play an important role in the different groups. From the inside to the outside, each circle in turn is phyla, class, order, family, genus level species; (f) LDA value distribution histogram. Different colors represent different groups of microorganisms that have a significant role. It mainly shows the significantly different species whose LDA score is greater than the preset value, namely the Biomaker with statistical difference, the default preset value is 2.0 (only the absolute value of the LDA value greater than 2 are shown in the figure). The colors of the bar chart represent the respective groups, and the length represents the LDA score, which is the size of the impact of significantly different species between the groups.
Further analysis of the top 10 most abundant bacterial species in K-GT, K-1LP, and K-7LP revealed that Komagataeibacter hansenii was dominant in all three kombucha groups, accounting for 99.9% in K-GT, 94.1% in K-1LP, and 84.7% in K-7LP. Compared to K-GT, the abundances of the other nine bacterial species were higher in K-1LP and K-7LP. These species included Megamonas rupellensis, Phascolarctobacterium faecium, Faecalibacterium prausnitzii, Prevotella copri, Ruminococcus faecis, Blautia wexlerae, Clostridium nexile, and Bacteroides faecichinchillae, as depicted in Fig. 1d. Notably, F. prausnitzii exhibited significant differences with proportions of 0%, 0.053%, and 1.13% in K-GT, K-1LP, and K-7LP, respectively (p = 0.046). The findings suggest that several of these bacteria, such as P. faecium, F. prausnitzii, and B. wexlerae, are beneficial to gut microbiota.
Additionally, based on the LEfSe analysis (Fig. 1e, f), Romboutsia was identified as a significantly different genus in K-1LP, while Faecalibacterium, Actinobacteria, and Bacilli were marked as significantly different species in K-7LP. It is evident that K-7LP displayed the highest bacterial abundance and diversity.
Analysis of ITS results for kombucha beverage liquid
-
Analysis of fungal OTUs (Fig. 2a, b) identified 26 fungal OTUs shared among the three groups. K-GT contained 82 unique OTUs, K-1LP had 31 unique OTUs, and K-7LP had 23 unique OTUs. These results indicated that K-1LP and K-7LP exhibited lower fungal diversity compared to K-GT (Fig. 2c).
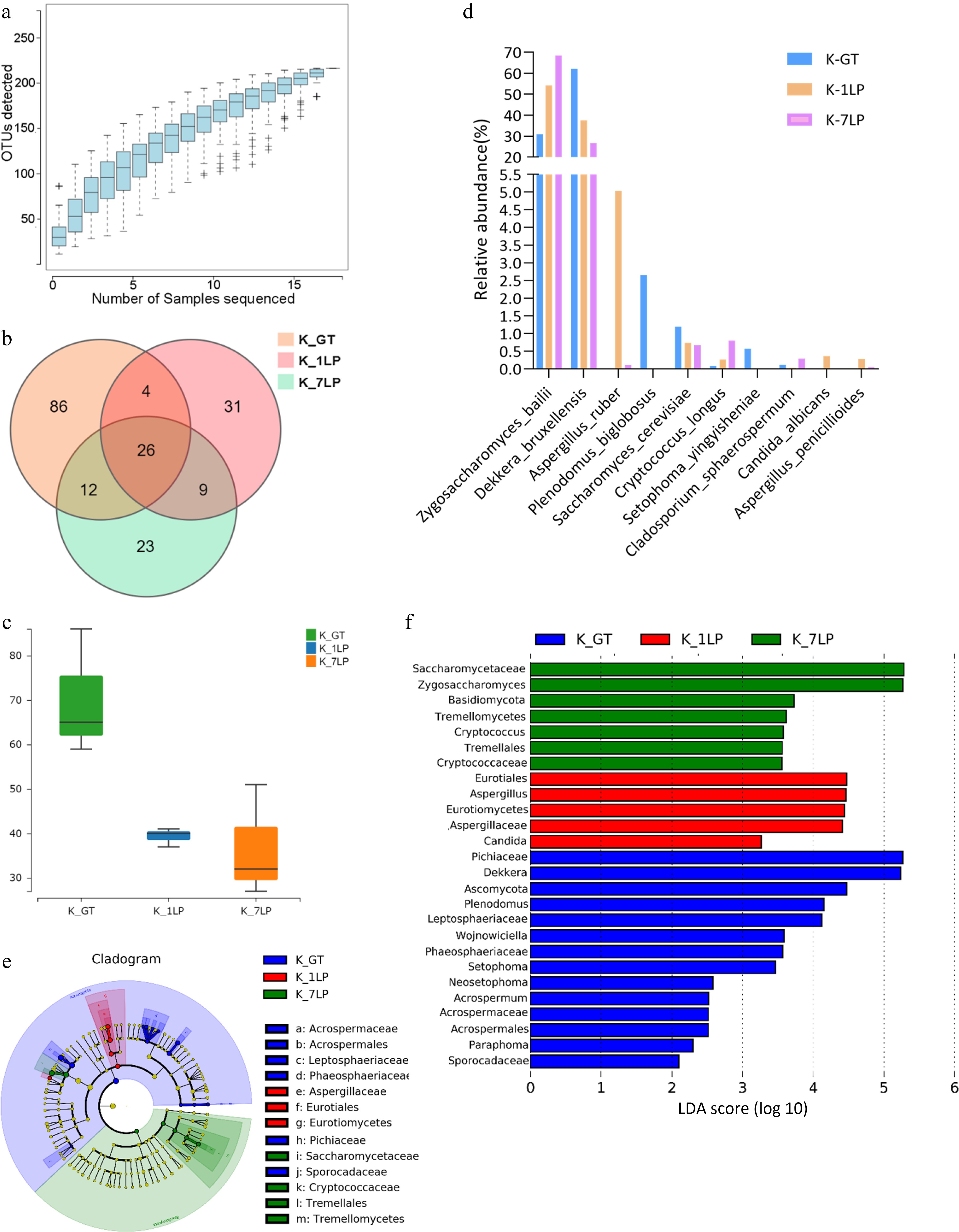
Figure 2.
Statistical analysis of ITS sequencing data for the three groups of kombucha beverages. (a) Fungal rarefaction curve; (b) Venn diagram of fungal OTUs; (c) Boxplot of α-diversity (sobs index). The Kruskal test is used to show differences in alpha diversity between groups. The five lines from bottom to top are: minimum, first quartile, median, third quartile, and maximum; (d) Histogram of relative abundance of three kinds of kombucha key fungal differences; (e) Evolutionary branch diagram. Different colors indicate different groups, and nodes in different colors indicate microbiomes that play an important role in the group represented by the color. A color circle represents a biomarker, and the legend in the upper right corner is the biomarker name. The yellow nodes represent microbial groups that do not play an important role in the different groups. From the inside to the outside, each circle in turn is phyla, class, order, family, genus level species; (f) LDA value distribution histogram. Different colors represent different groups of microorganisms that have a significant role. It mainly shows the significantly different species whose LDA score is greater than the preset value, namely the Biomaker with statistical difference, the default preset value is 2.0 (only the absolute value of the LDA value greater than 2 are displayed in the figure). The colors of the bar chart represent the respective groups, and the length represents the LDA score, which is the size of the impact of significantly different species between the groups.
Further analysis of the top 10 most abundant fungal species in K-GT, K-1LP, and K-7LP (Fig. 2d) revealed significant differences (p ≤ 0.05) among species such as Zygosaccharomyces bailii, Dekkera bruxellensis, Plenodomus biglobosus, Cryptococcus longus, Setophoma yingyisheniae, and Cladosporium sphaerospermum. D. bruxellensis was the predominant species in K-GT, accounting for 61.8% in K-GT, 37.5% in K-1LP, and 26.8% in K-7LP. Z. bailii was the dominant in K-1LP and K-7LP, making up 31.2%, 54.2%, and 68.4% in K-GT, K-1LP, and K-7LP, respectively.
LEfSe analysis of fungal classification highlighted the characteristic fungi at different taxonomic levels (Fig. 2e, f). In the K-GT, characteristic fungi included Pichiaceae, Dekkera, Ascomycota, Plenodomus, and Leptosphaeriaceae. For K-1LP, the characteristic fungi were Eurotiales, Eurotiomycetes, and Ascomycota. In K-7LP, the characteristic fungi were Saccharomycetaceae and Zygosaccharomyces.
Results of widely targeted metabolomics analysis
-
Following sensory evaluation and microbial detection of kombucha samples, K-GT and K-7LP showed higher quality and were therefore selected for UPLC-MS/MS analysis of tea metabolites. The OPLS-DA score plot (Fig. 3a) showed R2X at 89.6%, R2Y at 27.6%, indicating a close fit and distinct separation trends in the metabolic profiles among the groups, confirming that the sample data was reliable for further analysis. And the model was subjected to 200 permutation tests to verify its ability to classify correctly (Fig. 3b).
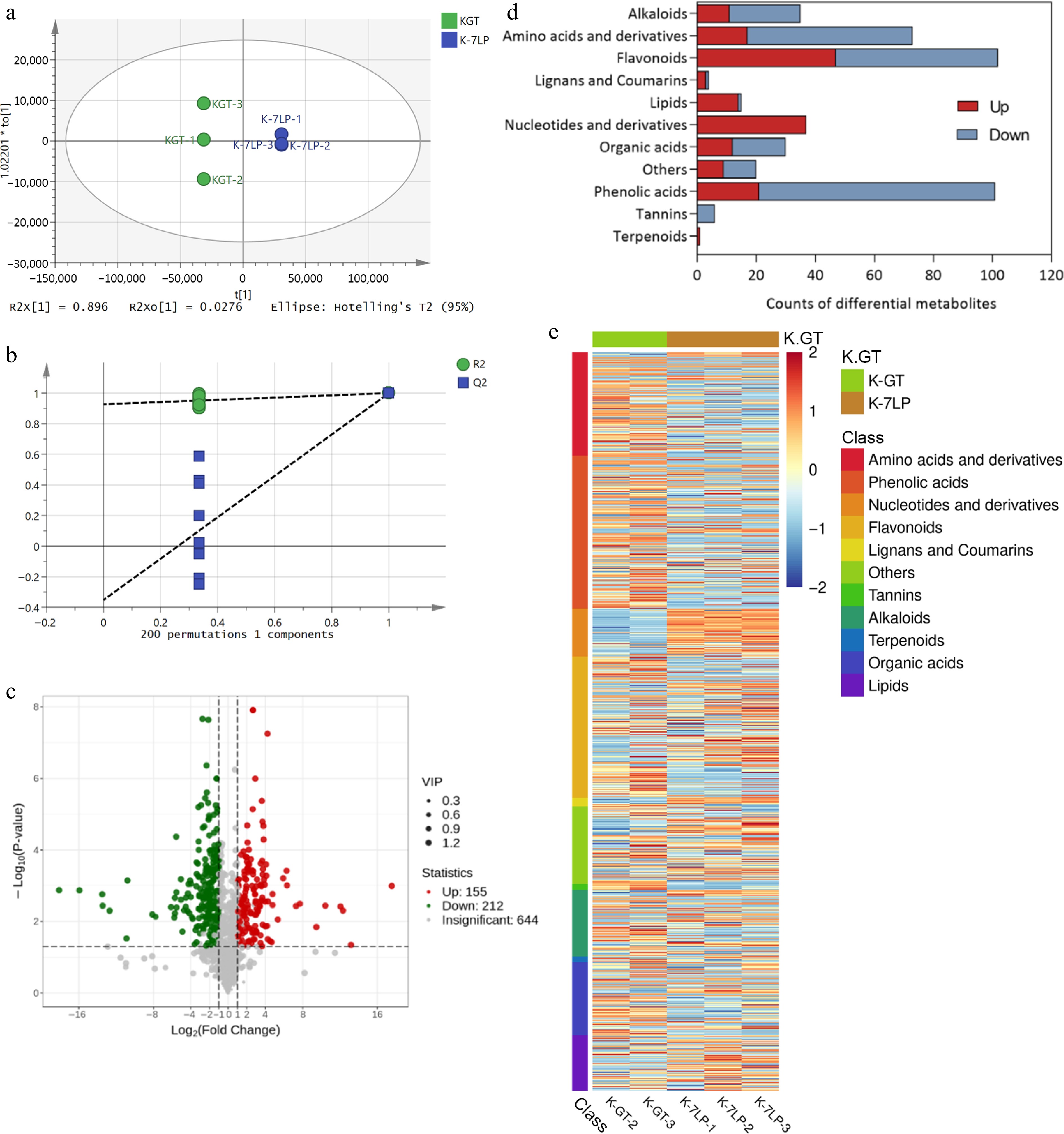
Figure 3.
Analysis of differential metabolites in K-GT and K-7LP. (a) OPLS-DA analysis; (b)Permutations plot for OPLS-DA model; (c) Volcano plot of differential metabolites; (d) Bar plot of differential metabolite classification; (e) Heatmap of differential metabolite abundance clustering.
Out of the 1,011 metabolites detected, 424 differential metabolites were identified based on a fold change of ≥ 2 or ≤ 0.5 between K-GT and K-7LP (Fig. 3c). In K-GT, the increased metabolites included 80 primary metabolites (56 amino acids and derivatives, 18 organic acids, and one lipid) and 174 secondary metabolites (80 phenolic acids, 55 flavonoids, and 24 alkaloids). In contrast, K-7LP showed an increase in 51 primary metabolites (37 nucleotides and derivatives, 17 amino acids and derivatives, 12 organic acids, and 14 lipids) and 82 secondary metabolites (47 flavonoids, 21 phenolic acids, and 11 alkaloids).
Heatmap analysis of the differential metabolites revealed that the differentially accumulated metabolites in K-GT were predominantly enriched in secondary metabolites, such as phenolic acids, amino acids, and derivatives, alkaloids, and tannins, whereas those in K-7LP were primarily enriched in primary metabolites, including nucleotides and derivatives, as well as lipids (Fig. 3e).
KEGG enrichment analysis was performed to determine the function of the differential metabolites in K-GT and K-7LP (Fig. 4a, b). The functional clustering analysis of differential metabolites using KEGG pathway analysis revealed that the differential metabolites in K-GT were enriched in 69 pathways. Among them, pathways such as aminoacyl-tRNA biosynthesis, arginine and proline metabolism, D-amino acid metabolism, arginine biosynthesis, and biosynthesis of amino acids were significantly enriched. A total of 25 differential metabolites, predominantly L-glutamine, L-arginine, L-aspartic acid, and N-α-acetyl-L-ornithine, were identified in the top five pathways, showing notable enrichment in these pathways. Amino acids such as glutamine, proline, histidine, arginine, and ornithine have the capability to transform into glutamic acid. These metabolites are primarily essential amino acids required for normal cell and tissue growth, and they play important roles in maintaining normal cellular metabolic processes[19]. The differential metabolites in K-7LP were enriched in 54 pathways. Significant enrichment was observed in pathways such as nucleotide metabolism, purine metabolism, vitamin B6 metabolism, caffeine metabolism, and pyrimidine metabolism. A total of 24 differential metabolites were involved in these five metabolic pathways, predominantly including xanthine, hypoxanthine, cytidine, and uridine, among other nucleotides and derivatives, which played a dominant role. This indicates that there were significant changes in caffeine metabolism in K-7LP, where caffeine is first metabolized into 1,7-dimethylxanthine, theobromine, and theophylline, and further metabolized into various xanthines and uric acid. Hypoxanthine, guanine, guanosine, and inosine are produced during purine degradation, and they are then converted into xanthine through the action of enzymes such as xanthine oxidase. The accumulation of these compounds can lead to increased antioxidant activity and other biological effects[20].
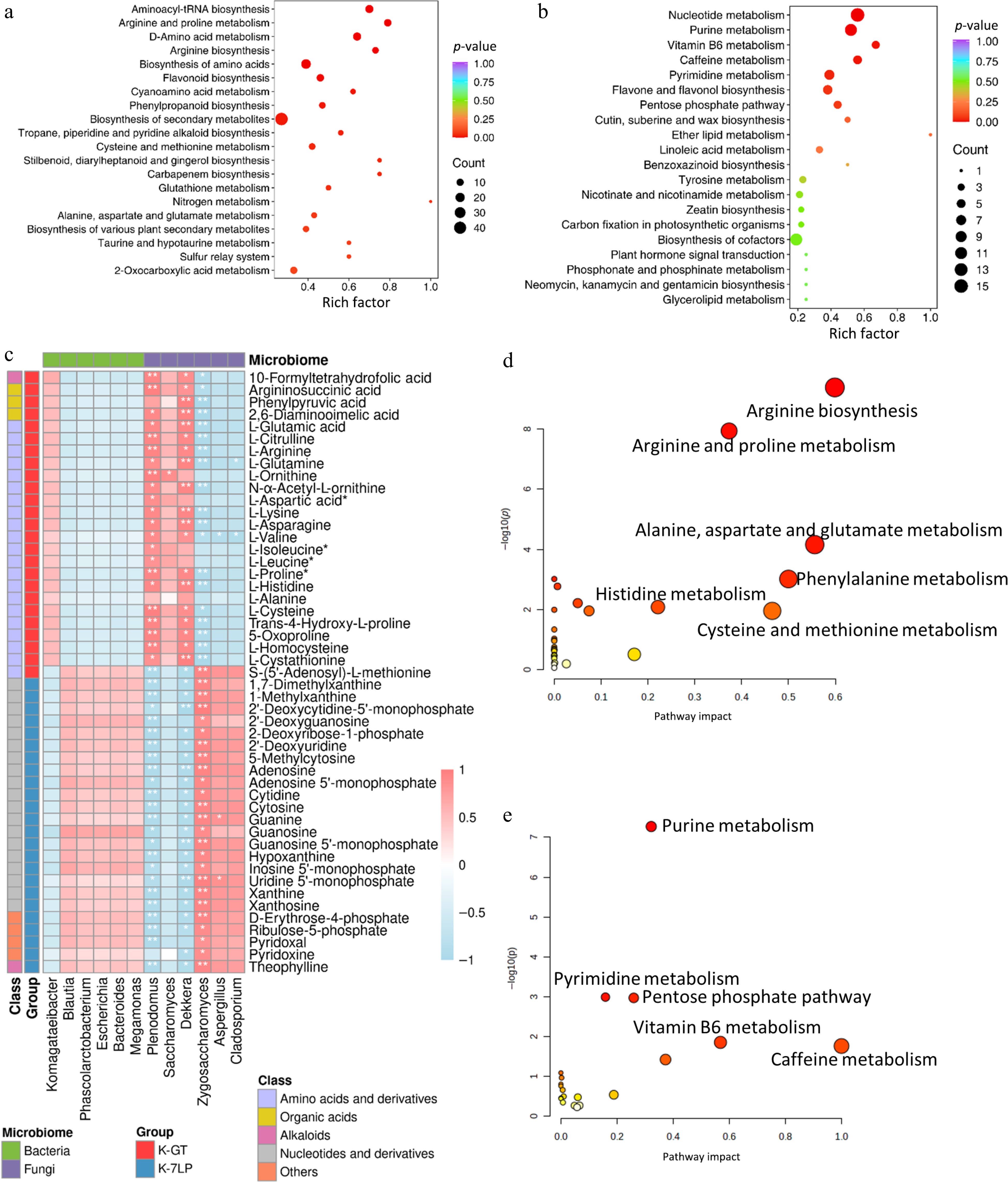
Figure 4.
KEGG enrichment analysis of differential metabolites in two types of kombucha. (a) KEGG enrichment analysis for differential metabolites in K-GT. (b) KEGG enrichment analysis for differential metabolites in K-7LP. Each bubble depicted in the graph represents a metabolic pathway; larger bubbles indicate a higher concentration of metabolites; the color of the bubble corresponds to the p-value in the enrichment analysis, with darker colors indicating a higher degree of enrichment. (c) Correlation analysis between key pathway active components and microbes: Pearson correlation analysis was conducted between 12 bacterial and fungal genera and 49 metabolites differentially enriched across 10 KEGG pathways. The horizontal axis represents the bacterial and fungal genera, while the vertical axis represents the 49 differentially enriched metabolites from these pathways. The colors red and blue indicate the Pearson correlation coefficient (r), with * and ** denoting significance levels of 0.01 < p < 0.05 and 0.001 < p < 0.01, respectively. (d) and (e) respectively show the scatter plots of clustering analysis of differential bioactive components for K-GT and K-7LP. In these plots, the X-axis represents the pathway impact, the Y-axis represents −log(p) value, the size of the circles represents the gene number, and the color represents the p-value, with darker colors indicating smaller p-values.
Correlation analysis between microorganisms and metabolites
-
Using Z-score normalization, the correlation analysis focused on 49 metabolites that were significantly linked to the top five pathways identified. The association between these 49 metabolites and the top 12 fungal and bacterial genera with high relative abundance based on OTU numbers were analyzed (Fig. 4c). The analysis revealed significant correlations between several fungal genera, including Dekkera, Plenodomus, Cryptococcus, Zygosaccharomyces, Cladosporium, and Aspergillus, and the majority of these metabolites.
In the context of K-GT, bacterial genera such as Saccharomyces, Dekkera, Setophoma, and Plenodomus were found to have positive correlation with the metabolites associated with the pathways. Conversely, the remaining nine bacterial genera demonstrated positive correlation with the pathway metabolites identified in K-7LP, highlighting the distinct microbial interactions within the two kombucha groups.
The clustering analysis (Fig. 4d, e) of differential bioactive components shows that the key metabolic pathways mainly affected in K-GT include arginine biosynthesis, arginine and proline metabolism, alanine, aspartate and glutamate metabolism, while those in K-7LP include purine metabolism, caffeine metabolism and vitamin B6 metabolism.
Differences in nucleotides and their derivatives between K-GT and K-7LP
-
Significant differences in nucleotides and their derivatives were observed between K-GT and K-7LP. In K-7LP, the concentrations of nucleotides and derivatives were in the following order: 8-Azaguanine, xanthine, hypoxanthine, allopurinol, and cytidine. Notably, nucleotides such as riboflavin 5'-adenosine diphosphate, allopurinol, and hypoxanthine exhibited significant increases in K-7LP. Riboflavin 5'-adenosine diphosphate was not detected at all in K-GT, while the levels of allopurinol and hypoxanthine in K-7LP were 207.02 times and 155.58 times higher than in K-GT, respectively. Riboflavin and its derivatives are water-soluble, essential vitamins and cofactors for enzymatic reactions, including those involving pyridoxal, pyridoxamine, pyridoxine, and their 5'-phosphates[21−23]. These compounds are involved in converting tryptophan to niacin, thereby activating vitamin B6. Vitamin B6 is crucial for the metabolism and conversion of carbohydrates, lipids, amino acids, and nucleic acids, making it an essential molecule for human health and normal functioning.
Differences in alkaloids between K-GT and K-7LP
-
In K-7LP, several alkaloid metabolites, including spermidine, isoquinoline, phenylethanolamine, 2,4,6,6-tetramethyl-3(6H)-pyridinone, and theophylline, were found at higher levels compared to K-GT. Putrescine and isoquinoline were not detected in K-GT, while phenylethanolamine, 2,4,6,6-tetramethyl-3(6H)-pyridinone, and theophylline were present at levels 79.90, 77.34, and 60.35 times higher in K-7LP, respectively. Research indicates that dietary spermidine can enhance health, improve learning and memory, and extend lifespan. Both spermidine and isoquinoline are found in many natural plants and exhibit a range of biological activities, including anti-inflammatory, anti-tumour, antimicrobial, and neuroprotective effects[24−26]. Additionally, theophylline, a component commonly found in tea, has been demonstrated to support weight-loss and lower lipid levels, thereby improving blood lipid profiles[27].
Differences in polyphenols between K-GT and K-7LP
-
Polyphenols are categorized into phenolic acids, flavonoids, polyphenol amides, lignans, and tannins, based on the number and arrangement of hydroxyl groups[28]. In K-7LP, phenolic acids such as salicylic acid, 2,3-dihydroxybenzoic acid, 3,4-dihydroxybenzoic acid, 2,5-dihydroxybenzoic acid, 2,4-dihydroxybenzoic acid, and 3,4-dihydroxyphenylacetic acid were upregulated, showing levels 5.46, 4.5, 4.5, 1.41, and 1.11 times higher than those in K-GT, respectively. These low molecular weight phenolic acids, typically produced through degradation and metabolism by the gut microbiota, possess a wide range of biological properties[29,30]. Due to their simple structure, they are more readily absorbed and exhibit higher bioavailability[31−33].
The flavonoid compounds detected were further categorized into flavones, chalcones, dihydroflavones, flavanols, dihydroflavonols, other flavonoids, and flavonols. K-GT contained 56 flavonoid metabolites, including 13 flavonols, 11 flavanols, 10 flavones, and 10 dihydroflavones. Conversely, the K-7LP group had 47 flavonoid metabolites, comprising 21 flavonols, 17 flavones, and three flavanols, with flavonols significantly increased in K-7LP. Notably, the increase in quercetin derivatives such as quercetin-3-O-(2''-O-rhamnosyl) galactoside and chrysoeriol-8-C-arabinoside-7-O-rutinoside were metabolites unique to K-7LP, highlighting the distinct phytochemical profile of this kombucha variant.
Differences in lipids between K-GT and K-7LP
-
Increasing evidence suggests that free fatty acids (FFAs) play a broad spectrum of roles, including the protection and restoration of tissue functionality, as well as the regulation of metabolism[34]. In comparison to K-GT, the lipid profile of K-7LP showed a notable enhancement, including 11 free fatty acids, two lyso-phosphatidylcholines, and one phosphatidylcholine. Notably, LysoPC 18:3 (2n isomer) was absent in K-GT, while the concentrations of 9,10,18-trihydroxystearic acid, 9,12,13-trihydroxy-10,15-octadecadienoic acid, and 10,16-dihydroxypalmitic acid in K-7LP were found to be 19.33, 19.14, and 8.60 times higher, respectively, compared to their levels in K-GT.
Differences in amino acids and their derivatives between K-GT and K-7LP
-
In K-7LP, cyclic dipeptides such as Cyclo(D-Leu-L-Pro), Cyclo(Pro-Pro), Cyclo(Ser-Pro) were significantly elevated. Amino acids and their derivatives, including L-Valyl-L-Leucine, Cyclo(D-Leu-L-Pro), Cyclo(Pro-Pro), and Cyclo(Ser-Pro), were identified with concentrations 704.81, 15.30, 15.30, and 12.94 times higher, respectively, than those found in K-GT. These cyclic dipeptide compounds are recognized for their ability to induce apoptosis in cancer cells, inhibit toxins, and exhibit anti-inflammatory and analgesic properties. As a distinct category of peptide compounds, cyclic dipeptides hold substantial potential for research and development due to their diverse biological activities. They are particularly noted for their roles in promoting apoptosis in cancer cells, inhibiting toxins, and providing anti-inflammatory and analgesic benefits, making them a prominently researched class of compounds with significant potential for further investigation and application in the realm of biological activities[35].
Network pharmacology analysis
-
A network diagram illustrating the interaction between active components and target genes was established using Cytoscape 3.7.1 (Fig. 5a, b).
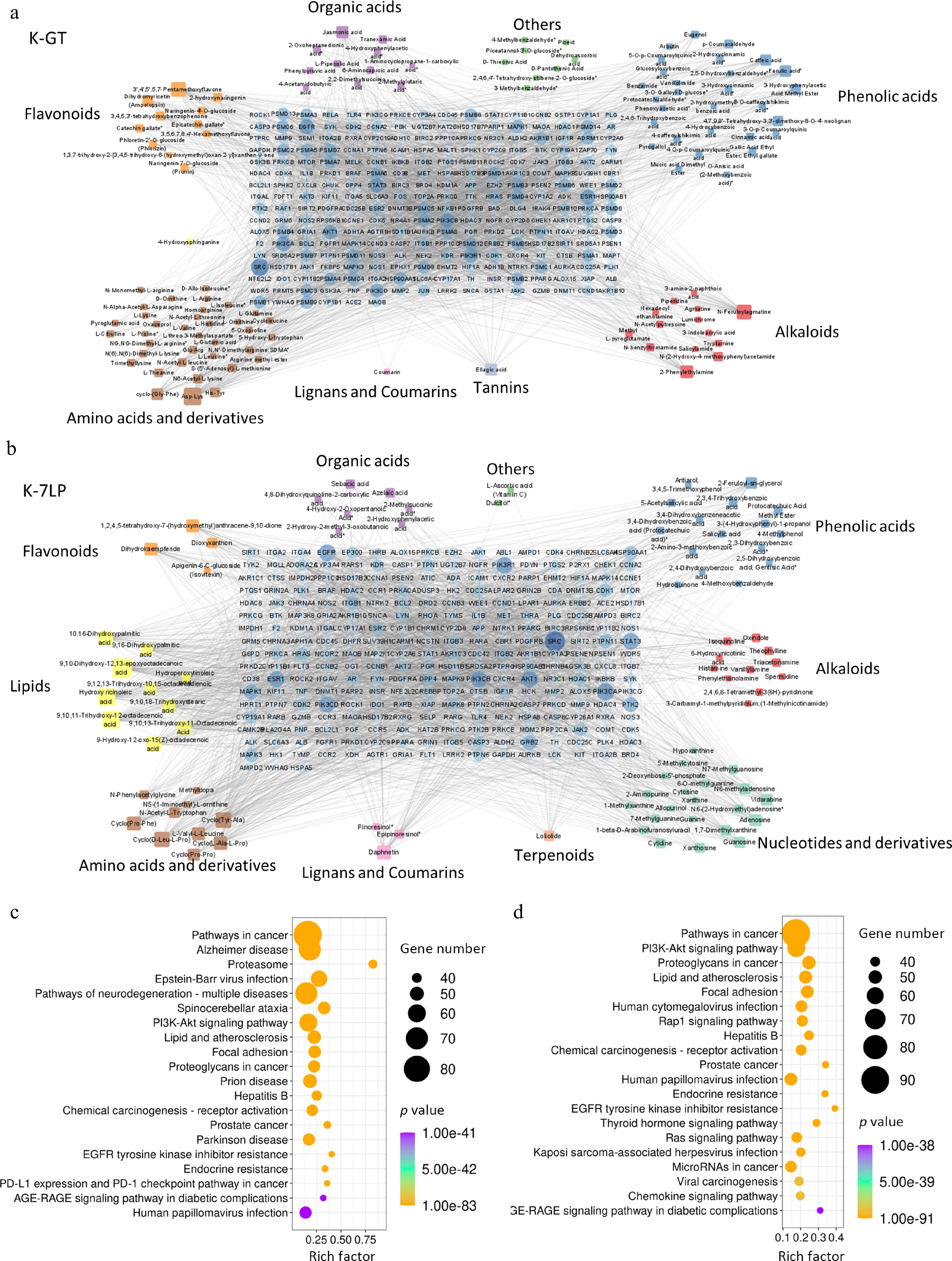
Figure 5.
Key bioactive components, key target genes, and PPI network diagrams for (a) K-GT, and (b) K-7LP. In the PPI network diagrams, circles represent target genes, with their size and color intensity indicating the degree of connectivity to active components and their importance in the network. Squares represent active components, with their size and color depth proportional to the number of connected target genes. The network topology analysis based on connectivity determined the core bioactive components and key target genes for K-GT and K-7LP. (c) and (d) respectively show the KEGG enrichment analysis scatter plots of target genes affected by differential bioactive components for K-GT and K-7LP. In these plots, the X-axis represents the rich factor, the Y-axis represents the pathway names, the size of the circles represents the gene number, and the color represents the p-value, with darker colors indicating smaller p-values.
In K-GT, 262 nodes exceeded the average node degree (6.74), resulting in 374 targets and 3,042 edges being generated in Cytoscape. Additionally, 112 active components were associated with the target, including 36 types of amino acids and their derivatives, 30 types of phenolic acids, 14 types of alkaloids, and 11 types of flavonoids. Based on the degree of connection, the top three active ingredients in K-GT were Asp-Lys, 3',4',5',5,7-pentamethoxyflavone, and N-Feruloylagmatine. In K-7LP, there were a total of 275 nodes that exceeded the average node degree (5.51), generating a total of 360 targets and 2,781 edges. Furthermore, there were a total of 85 active components associated with the target in this group which included: 21 nucleotides and their derivatives, 16 phenolic acids, 11 alkaloids, and 10 lipids. According to their degree of connection within this group; the top three active ingredients identified are: 9,10-dihydroxy-12,13-epoxyoctadecanoic acid, 9,10,11-trihydroxy-12-octadecenoic acid, and hydroxy ricinoleic acid. Compared with K-GT, the active components of lipids, and nucleotides, and derivatives in K-7LP were significantly increased.
KEGG enrichment analysis of the target genes revealed that the K-GT targets were significantly enriched in pathways such as cancer, alzheimer's disease, and proteasome (Fig. 5c, d). K-7LP was mainly enriched in pathways such as cancer, the PI3K-Akt signaling pathway, and the pathway involving proteoglycans in cancer.
-
There is a marked distinction in the bacterial community structure between K-GT and K-7LP. K-7LP exhibits a greater bacterial diversity than K-GT, including beneficial bacterial genera such as Faecalibacterium, Bacteroides, Blautia, and Clostridium XlVa. These bacteria play significant roles in regulating intestinal motility, enhancing intestinal barrier functions, and mitigating intestinal diseases[36]. Notable bacterial species identified include F. prausnitzii, B. wexlerae, and Prevotella copri. Phascolarctobacterium is particularly significant as a producer of acetate and propionate. Notably, F. prausnitzii, which is considerably more abundant in K-7LP, ferments glucose to produce acetate, butyrate, d-lactate, and formate, making it a key butyrate-producing bacterium in the human colon[37]. Furthermore, the anti-inflammatory metabolite salicylic acid, produced by F. prausnitzii, was found in K-7LP at levels 5.46 times higher than in K-GT. B. wexlerae, Prevotella copri, and P. faecium, prevalent in the human gastrointestinal tract, are deemed beneficial for their ability to generate short-chain fatty acids[38−40]. Clostridium and Eubacterium, major bacterial genera involved in the metabolism of phenolic compounds, can produce low-molecular-weight polyphenols, enhancing bioavailability[41]. On the fungal front, K-7LP significantly surpasses K-GT with an increase in Z. bailii, a yeast commonly found in various food fermentations with potential for producing lactic acid and ethanol.
Currently, many nucleotides and their derivatives possess antifungal properties and are being developed into new antifungal medications to treat fungal infections in humans and plants[42,43]. This could explain the lower fungal diversity observed in K-7LP compared to K-GT. Caffeine, theobromine, and theophylline, the primary purine alkaloids found in tea, are significantly degraded fungi such as Aspergillus in K-7LP[44]. Along with other microbial populations, these fungi facilitate the metabolism of 1,7-dimethylxanthine and theophylline into compounds such as xanthine, hypoxanthine, and guanosine.
-
This study highlights that dark tea kombucha, as a unique tea beverage, is abundant in microorganisms and chemical components. The findings suggest that beneficial gut microbes, including F. prausnitzii and M. rupellensis, were identified, along with the abundance of bioactive components such as nucleotides and their derivatives, lipids, phenolic acids, and flavonoids. These may offer potential and prospects for the development of products with physiological activity. The microbial fermentation process endows this tea with distinctive characteristics and health benefits, thereby increasing its attractiveness and popularity in the market. Given the growing consumer interest in healthy diets and natural products, dark tea kombucha, as a health beverage with multiple physiological activities, stands to gain greater development and utilization value. Thus, research into dark tea kombucha will enhance our understanding of its mechanisms and effects, fostering innovative ideas and product options for the future kombucha industry.
This research was funded by the Guangxi Innovation Driven Development Special Fund Project (Grant No. AA20302018), the National Key R&D Program of China (2018YFC1604405), the National Tea Industry Technology System Research Project of China (CARS-19-C01), the Key R&D Program of Hunan Province (2020WK2017), National Natural Science Foundation Project of China (31471590, 31100501), and the Self-Science Foundation of Hunan Province, China (2019jj50237).
-
The authors confirm contribution to the paper as follows: performing experiments, data curation, writing the original draft: Nie Q; investigation, visualization: Guo Q, Ding S, Xie M, Wu H, Yuan J, Pang Y, Liao X, Liu Z; resources, funding acquisition, supervision, writing–review: Liu Z, Cai S. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article.
-
The authors declare that they have no conflict of interest.
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Nie Q, Ding S, Xie M, Wu H, Guo Q, et al. 2025. Exploring the microbial composition and metabolites in dark tea Kombucha: a comparative analysis with green tea Kombucha. Beverage Plant Research 5: e002 doi: 10.48130/bpr-0024-0038 

Exploring the microbial composition and metabolites in dark tea Kombucha: a comparative analysis with green tea Kombucha
- Received: 14 September 2024
- Revised: 17 October 2024
- Accepted: 05 November 2024
- Published online: 17 January 2025
Abstract: Kombucha is typically fermented from green or black tea using symbiotic colonies of bacteria and yeasts (SCOBY), known for its rich bioactive components. However, dark tea kombucha, particularly its nonvolatile metabolites, remains underexplored. This study compares the microbial composition and active components of green tea kombucha (K-GT) with those of a 7-year-old Liupao tea kombucha (K-7LP). Both kombuchas were free of pathogenic bacteria, with K-7LP exhibiting greater bacterial diversity, including beneficial species such as Faecalibacterium prausnitzii, Blautia wexlerae, and Phascolarctobacterium faecium. The dominant fungi in K-GT were Dekkera bruxellensis, whereas K-7LP primarily contained Zygosaccharomyces bailii, accounting for 61.8% in K-GT, and 26.8% in K-7LP. Different metabolites between K-GT and K-7LP included 47 flavonoids, 37 nucleotides and derivatives, 21 phenolic acids, and 14 lipids. Notably, nucleotides such as adenosine, adenosine 5'-monophosphate, and 1,7-dimethylxanthine displayed significant associations with fungi like Aspergillus, Plenodomus, and Dekkera. Overall, compared to one-year-aged Liupao tea, seven-year-aged Liupao tea is a superior substrate for kombucha fermentation. Compared to traditional green tea kombucha, dark tea kombucha emerges as a superior beverage, characterized by its unique sweet taste, beneficial microorganisms, and high bioactivity, offering promising prospects and exploitability for its health benefits.
-
Key words:
- Dark tea kombucha /
- Liupao tea /
- 16S rRNA /
- ITS /
- UPLC-MS/MS /
- Aspergillus /
- Plenodomus /
- Nucleotides and derivatives