










-
The genus Nymphaea, belonging to the family Nymphaeaceae and commonly known as water lily, comprises over 60 species of perennial aquatic plants[1]. This precious aquatic plant is distributed across frigid zones to the tropical regions worldwide. In horticulture, water lilies can be divided into two categories based on their ecological habits: tropical water lilies and hardy water lilies[2]. In addition to their aesthetic appeal, economically, Nymphaea species possess a substantial quantity of phytochemicals and nutrients, making them widely utilized for beverage preparation[3], essential oil extraction[4], and as a valuable source of food, nutrition, and medicinal purposes[5,6]. The incorporation of water lily in scented tea offers potential benefits in the tea beverage industry, combining ornamental value and potential health benefits[7]. Understanding the chemical composition and metabolic profiles of water lilies is important for uncovering their biological functions and applications. Metabolomics, a comprehensive study of small molecule metabolites, provides insights into the diverse range of non-volatile and volatile metabolites in water lilies[8,9].
Non-volatile metabolites encompass a wide range of compounds, including primary metabolites such as carbohydrates, amino acids, and organic acids, as well as secondary metabolites such as flavonoids, alkaloids, and phenolic compounds[10]. These metabolites are essential for the growth, development, and defense mechanisms of plants[11]. Phytochemical screening of water lilies indicates the presence of several bioactive compounds like phenolic, flavonoids, triterpenes, glycosides, carbohydrates, and other compounds[12]. Phenols and flavonoids are the main phytochemicals found in water lilies and are responsible for their health benefits[5]. In addition, the captivating colors of water lily petals are attributed to their richanthocyanin content. For instance, delphinidin 3′-O-(2″-O-galloyl-6″-O-acetyl-β-galactopyranoside was identified as the primary blue anthocyanin in N. colorata[13]. Most detection methods for water lily metabolites are either targeted, with limited coverage, or non-targeted, with lower sensitivity and accuracy[14]. The combination of ultra-high-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) with widely targeted metabolomics techniques provides fast separation, high sensitivity, and broad coverage[15].
Volatile metabolites, low molecular weight compounds that evaporate at ambient temperatures, are essential for the aroma, pollinator attraction, communication, and defense in plants. Its compositions vary among different species of water lilies. For example, day-blooming species emitting aromatic alcohols and ethers, while nocturnal species have a higher abundance of aromatic ethers, aliphatic esters, and C5-branched chain esters, which may play a role in attracting potential pollinators through olfactory cues[16]. In addition, previous studies using solid phase microextraction gas chromatography-mass spectrometry (SPME-GC-MS) detected 79 and 71 volatile compounds in tropical water lilies and hardy water lilies, respectively, with aromatic substances being their major volatile components[17]. However, with the advancements in analytical techniques, researchers have sought to explore more advanced methods for volatile compound analysis. Currently, comprehensive two-dimensional gas chromatography-time-of-flight mass spectrometry (GC×GC-TOFMS) has been widely adopted for the analysis of volatile compounds in a variety of foods due to its high resolution, high sensitivity, and peak capacity[18,19].
In this study, we utilized UPLC-MS/MS and GC×GC-TOFMS techniques to comprehensively identify both non-volatile and volatile constituents of water lilies in five different colors. By comparing metabolite profiles across various Nymphaea species and varieties, we can uncover the discrepancies in chemical compounds that maybe linked to variations in scent, coloration, and potential biological activity. The findings of this study will enhance our understanding of the metabolic variability and chemical constitution of water lilies, thereby improving our knowledge of their distinctive attributes and potential uses.
-
Methanol, acetonitrile and ethanol were purchased from Merck (Darmstadt, Germany). Ether (GC) was acquired from Tedia (Fairfield, OH, USA). Anhydrous sodium sulfate (AR) and ethyl decanoate (AR, internal standard) were purchased from Sigma-Aldrich (Shanghai, China). Distilled water was obtained from Wahaha Group Company (Hangzhou, China). The carboxen/divinylbenzene/polydimethylsiloxane (CAR-DVB-PDMS; 50/30 μm) microextraction fiber was obtained from Supelco (Bellefonte, PA, USA). The n-alkanes (C3-C9, C8-C40) were purchased from J&K Scientific (Beijing, China).
Sample preparation
-
As shown in Fig. 1, five different water lily samples were collected from Hangzhou Aquatic Plant Society, including two species — N. lotus (NL), N. rubra (NR), and three varieties — Nymphaea 'Texas Dawn' (TD), Nymphaea 'Blue Bird' (BB), and Nymphaea 'Detective Erika' (DE). Three fully expanded flowers (on the first day after flowering) were collected in the morning, three samples of each variety, and processed immediately.
Figure 1.
Five different species and varieties of water lilies selected in this study.
Analysis of non-volatile metabolites
Sample pre-treatment
-
The petals of water lily samples were ground using a mixer mill (MM 400,) with a zirconia bead for 1.5 min at 30 Hz. One hundred mg of sample powder was dissolved with 0.6 mL 70% aqueous methanol; the dissolved sample was placed in the refrigerator at 4 °C for 12 h, during which the sample was vortexed six times; finally, the sample was centrifuged at 4 °C for 10 min at 12,000 r/min, and the extracts were absorbed and filtrated (SCAA-104, 0.22 μm pore size; ANPEL, Shanghai, China), and then sealed in the injection vial for subsequent UPLC-MS/MS analysis.
UPLC-MS/MS analysis
-
The analysis was conducted by MetWare (Wuhan, China)[20]. UPLC (Shim-pack UFLC SHIMADZU CBM30A) equipped with tandem mass spectrometry (Applied Biosystems 4500 QTRAP) was used for the wide-targeted metabolomic assays of non-volatiles in water lily samples. The analytical conditions were as follows: Waters ACQUITY UPLC HSS T3 C18 column (2.1 mm × 100.0 mm, 1.8 μm); flow rate of 0.35 mL/min; column temperature of 40 °C; injection volume of 4 μL. The mobile phase A was ultrapure water (dissolved in 0.04% acetic acid), and the mobile phase B was acetonitrile (dissolved in 0.04% acetic acid). The mobile phase elution gradient was as follows: the proportion of phase B was 5% at 0.00 min, the proportion of phase B increased linearly to 95% at 10.00 min and maintained at 95% for 1 min, the proportion of phase B was reduced to 5% at 11.00–11.10 min and equilibrated with the proportion of phase B at 5% for 14 min. The effluent was alternatively connected to an ESI-triple quadrupole-linear ion trap (QTRAP)-MS.
Linear ion trap (LIT) and triple quadrupole (QQQ) scans were acquired on a triple quadrupole-linear ion trap mass spectrometer (Q TRAP), API 4500 Q TRAP UPLC/MS/MS System, equipped with an ESI Turbo Ion-Spray interface, operating in positive and negative ion mode and controlled by Analyst 1.6.3 software (AB Sciex). The ESI source operation parameters were as follows: ion source, turbo spray; source temperature 550 °C; ion spray voltage (IS) 5,500 V (positive ion mode)/–4,500 V (negative ion mode); ion source gas I (GSI), gas II (GSII), curtain gas (CUR) were set at 50, 60, and 30 psi, respectively; the collision gas (CAD) was high. Instrument tuning and mass calibration were performed with 10 and 100 μmol/L polypropylene glycol solutions in QQQ and LIT modes, respectively. QQQ scans were acquired as MRM experiments with collision gas (nitrogen) set to 5 psi. Declustering potential (DP) and collision energy (CE) for individual MRM transitions was carried out with further DP and CE optimization. A specific set of MRM transitions were monitored for each period according to the metabolites eluted within this period.
Qualitative and quantitative analyse of non-volatiles
-
For the qualitative analysis, the metabolites were identified by matching the retention time, fragmentation patterns, and accurate m/z values to the standards in the self-constructed metabolite database (MetWare, Wuhan, China). The quantitative analysis was conducted based on the signal intensities of metabolites obtained from characteristic ions. MultiaQuant software was used to integrate and calibrate chromatographic peaks. The peak area of each chromatographic peak represented the relative content of the corresponding substance.
Analysis of volatile metabolites
Extraction of aroma components
-
The sample preparation method underwent minor modifications, as outlined in the previous study[19]. Specifically, simultaneous distillation extraction (SDE) method was used to extract the aroma components of water lily flower. The specific steps were as follows: weigh 10.00 g of the water lily flower sample to be tested, put it in a 500 mL round-bottomed flask and add 300 mL of boiling distilled water, and heat it to a slight boil with an electric heating jacket. Add 30 mL of redistilled anhydrous ether into the extraction flask and distill the extract at 50 °C for 1 h. Remove the water from the obtained material with anhydrous sodium sulfate and concentrate under nitrogen, then put into the injection bottle for sealing and storage at –20 °C for measurement.
GC×GC-TOFMS analysis conditions
-
The analysis of aroma components in the water lily samples was conducted using a GC×GC-TOFMS system, which consisted of a gas chromatograph (Agilent 7890B; Santa Clara, CA, USA) coupled with a TOFMS instrument (LECO Pegasus 4D; LECO Corporation, St. Joseph, MI, USA). The first dimension (1st D) column was a non-polar DB-5MS column (30 m × 250 μm × 0.25 μm) and the second dimension (2nd D) column was a moderate polar DB-17HT column (1.9 m × 100 μm × 0.10 μm), and both above columns were purchased from Agilent Technologies (Santa Clara, CA, USA). Injection port temperature: 280 °C; Transfer line temperature: 270 °C; Carrier gas: helium (purity 99.999%); No split injection; Modulation time interval: 4.0 s; Sample injection volume: 1.0 μL. The 1st D column temperature program: hold at 60 °C for 3.0 min, ramp at 4.0 °C/min to 280 °C, hold for 2.5 min; 2nd D column temperature program: hold at 65 °C for 3.0 min, ramp at 4.0 °C/min to 280 °C, hold for 2.5 min; Total analysis time: 60.5 min. MS conditions: Electron ionization source; Ionization energy: –70 eV; Mass scanning range: 33 to 600 m/z; Ion source temperature: 220 °C.
Qualitative analysis of aroma components
-
The GC×GC-TOFMS data were processed using the LECO ChromaTOF software. The calculated retention index (RI) values calculated using C8–C40 n-alkanes, and the reference RI values were obtained from the NIST 2014 semi-standard non-polar (DB-5) column. A compound was considered tentatively identified if the difference between the calculated and reference RI values was less than 20.
Statistical data analysis
-
Patrial least squares discriminant analysis (PLS-DA) was performed by MetaboAnalyst 5.0 (
www.metaboanalyst.ca ). The data was log transformed (log10) and normalization by median before PLS-DA. Significantly regulated metabolites between groups were determined by VIP ≥ 1 and absolute Log2FC (fold change) ≥ 1. To mitigate the risk of overfitting, we conducted a permutation test with 100 permutations.The identified non-volatile metabolites were annotated using Kyoto Encyclopedia of Genes and Genomes (KEGG) Compound database (
www.kegg.jp/kegg/compound ), annotated metabolites were then mapped to KEGG pathway database (www.kegg.jp/kegg/pathway.html ). Pathways with significantly regulated metabolites mapped to were then fed into metabolite sets enrichment analysis, their significance was determined using p-values obtained from the hypergeometric test. -
A total of 533 non-volatile metabolites including nine categories were tentatively identified for water lilies by a comparison with tandem mass spectrum information from published databases and standards from the MetWare self-constructed metabolite database (Supplemental Fig. S1). As shown in Fig. 2a, these were 151 flavonoids (including 64 flavonoid and flavonoid carbonoside, 51 flavonols, 11 anthocyanins, 11 flavanols, nine dihydroflavone and dihydroflavonol, five isoflavones), 109 phenolic acids, 68 amino acids and derivatives, 57 lipids, 39 nucleotides and derivatives, 30 organic acids, 27 alkaloids, 23 saccharides and alcohols, and 29 other metabolites. Results showed that flavonoids, phenolic acids, lipids, amino acids and derivatives were the dominant non-volatile metabolites in the five water lilies. To better understand the differences in the content of non-volatile components between water lily samples from different species and varieties, the analysis was calculated by Log2FC (fold change) ≥ 2 or ≤ 0.5. The plots reflect the information on differential metabolite up-regulation and down-regulation (Fig. 2b). The numbers of differential metabolites identified in NL, TD, NR, and DE were 329 (237 up, 92 down), 310 (218 up, 92 down), 314 (213 up, 101 down), and 282 (177 up, 105 down), respectively.
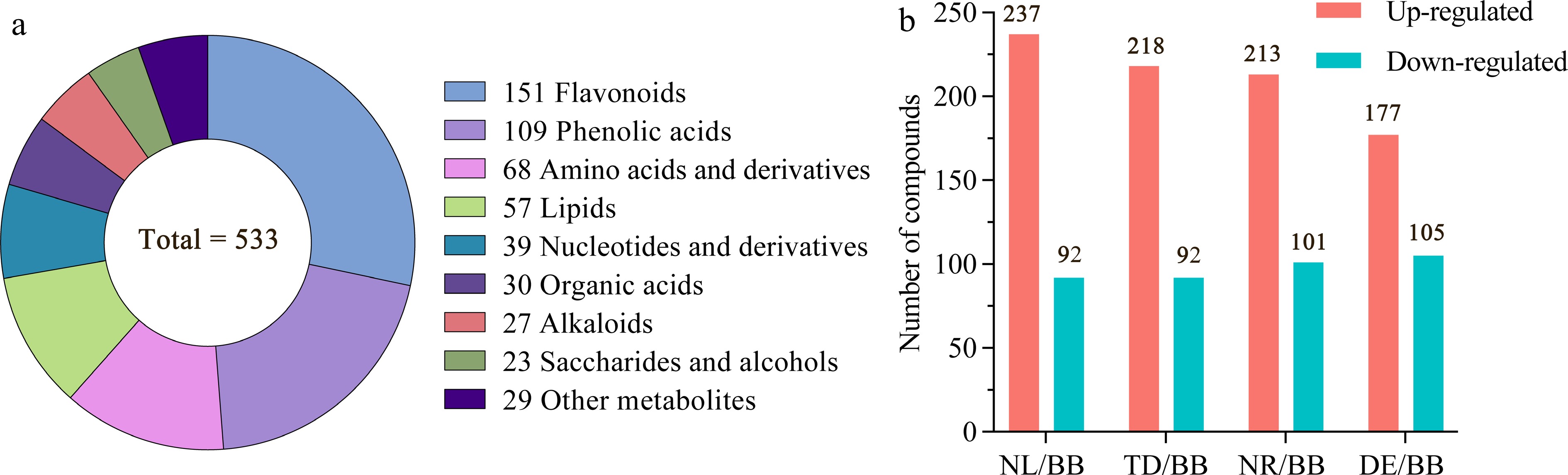
Figure 2.
Overview of the non-volatile components. (a) Quantitative distribution of chemical classes of volatile compounds. (b) Number of differentiated compounds with fold change ≥ 2 or ≤ 0.5. Note: NL, N. lotus; NR, N. rubra; TD, Nymphaea 'Texas Dawn'; BB, Nymphaea 'Blue Bird'; DE, Nymphaea 'Detective Erika'.
Crucial differential metabolites in water lilies
-
In the present study, all differential metabolites were investigated based on fold change, and a total of 118 non-volatiles were screened in five water lilies, mainly consisted of flavonoids, phenolic acids, amino acids and derivatives, lipids, and organic acids metabolites (Supplemental Table S1 & Supplemental Fig. S2). Specifically, we screened 10 up-regulated and 10 down-regulated metabolites with the highest fold change values in different water lilies (Fig. 3). Comparing the NL samples to the BB samples, we observed higher levels of chrysoeriol-O-malonylhexoside, 6,7,8-tetrahydroxy-5-methoxyflavone, myricetin-O-glucoside-rhamnoside, and kaempferol-3-O-neohesperidoside. On the other hand, the BB samples contained higher levels of isoquercitrin and luteolin-7-O-β-D-gentiobioside. Moreover, the TD samples exhibited high levels of neochlorogenic acid, chlorogenic acid, 1-O-p-coumaroylquinic acid, and tetragallic acid, while the BB samples exhibited high levels of 2'-hydroxygenistein, phloretin 2'-O-glucoside, and 3,4-dihydroxybenzaldehyde. Comparing the NR samples to the BB samples, we found higher levels of cyanidin-3-rutinoside, cyanidin-3-O-galactoside, and myricetin-O-glucoside-rhamnoside. Conversely, the BB samples exhibited higher levels of 7-methoxycoumarin, myricitrin, and naringenin-7-O-glucoside. In comparison to the BB samples, the DE samples displayed higher levels of myricetin-O-glucoside-rhamnoside, cis-4-hydroxy-D-proline, myricetin-3-O-rhamnoside-7-O-rhamnoside, and neochlorogenic acid. On the other hand, the BB samples contained higher levels of myricitrin, isoquercitrin, and naringenin-7-O-glucoside.
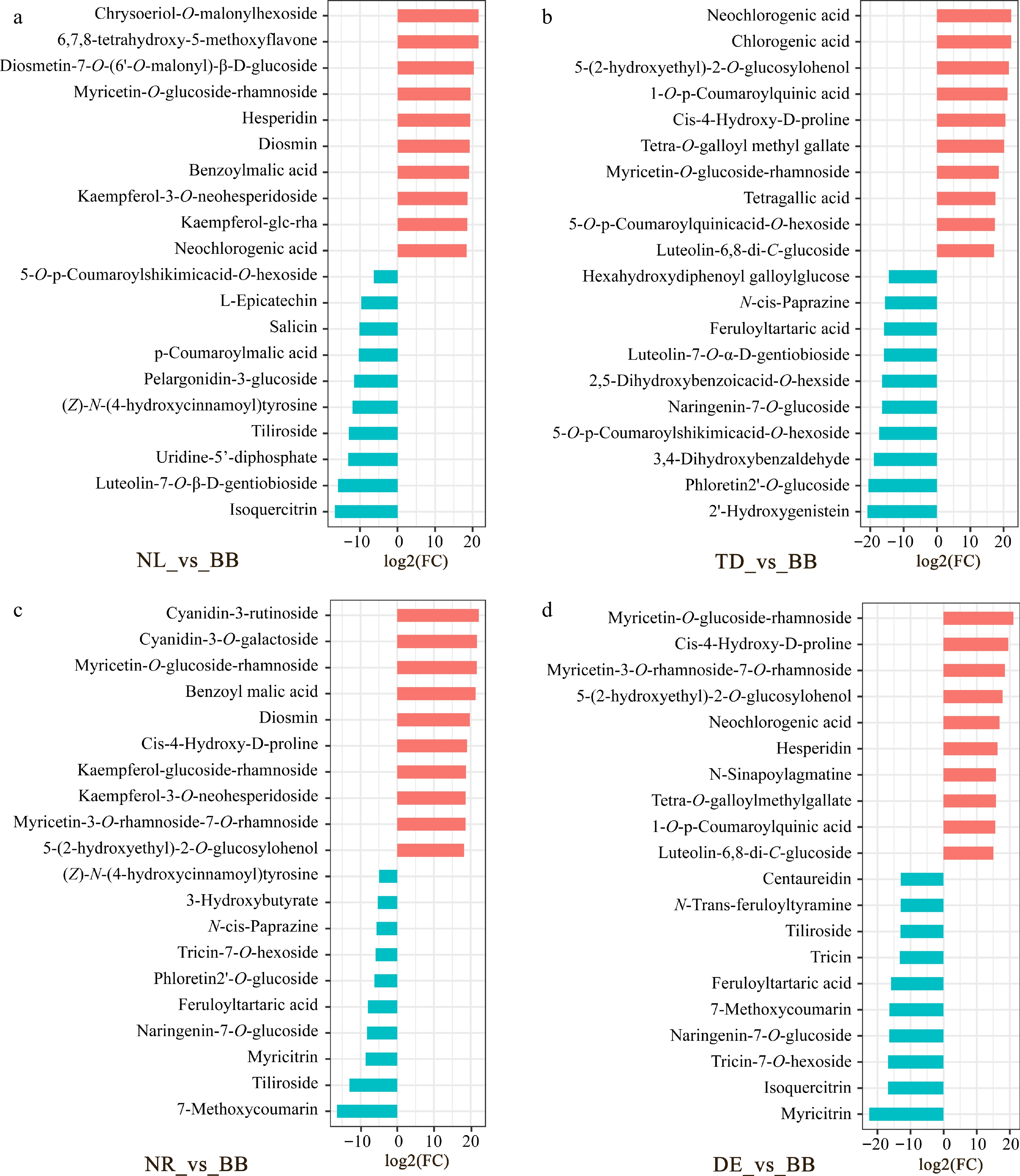
Figure 3.
The highest fold change values of non-volatile metabolites. (a) Fold change plot of NL vs BB. (b) Fold change plot of TD vs BB. (c) Fold change plot of NR vs BB. (d) Fold change plot of DE vs BB. Note: The horizontal coordinate is the log2FC of the differentially metabolized metabolite, and the vertical coordinate is the differentially metabolized metabolite. Red represents up-regulated differentially expressed metabolites, cyan represents down-regulated differentially expressed metabolites. NL, N. lotus; NR, N. rubra; TD, Nymphaea 'Texas Dawn'; BB, Nymphaea 'Blue Bird'; DE, Nymphaea 'Detective Erika'.
We further identified the enrichment of differential metabolites in the KEGG mapping. The results of pathway enrichment analysis of the detected differential compounds using the KEGG database are shown in Supplemental Fig. S3. A total of 354, 327, 380, 299 differential compounds from BB vs NL, BB vs TD, BB vs NR, BB vs DE samples could be annotated to the relevant metabolic pathways, which were mainly significantly enriched in the pathways of biosynthesis of secondary metabolites, flavonoid biosynthesis, anthocyanin biosynthesis, flavonoids and flavonols biosynthesis, isoflavonoid biosynthesis. Furthermore, we observed significant enrichment in pathways associated with tryptophan metabolism and phenylpropanoid biosynthesis in the BB vs TD group. Additionally, caffeine metabolism exhibited significant enrichment in the BB vs NR group.
Identification and overview of volatile compounds
-
The fragrance of water lily contains volatile compounds such as terpenes, phenylpropanoids, benzenoids, fatty acid derivatives, and amino acid derivatives. These compounds not only attract pollinators, but also play a crucial role in transmitting signals in plant-plant interactions and providing protection and defense for the plant[17]. In this study, the volatile compounds of water lily samples were analyzed by GC×GC-TOFMS (Supplemental Fig. S4). By comparing the MS of the compounds and comparing the chromatographic peaks and tested RI values with the reported RI values, 166 volatiles were identified, including 46 aromatic compounds, 34 alkynes, 22 ketones, 10 alcohols, 18 esters, 20 aldehydes, three carboxylic acids, five heterocyclics, five sulfur-containing compounds, and three other compounds (Fig. 4a). We identified 128, 141, 142, 135, and 129 volatile compounds in BB, NL, TD, NR, and DE, respectively, and 108 of the 166 metabolites were common to all water lily samples (Fig. 4b). We observed that 20 volatile compounds were exclusively detected in specific water lily samples. For instance, in BB variety, compounds such as amorpha-4,11-diene, (Z)-geranylacetone, α-bisabolol, and (E)-β-ionone were found. In NR variety, pyrocinchonic anhydride and 2-tetradecanone were exclusively detected. NL variety exhibited unique compounds including mequinol, 4-ethylresorcinol, p-xylene, and 2,5-hexanedione. TD variety showed the presence of terpilene, (E,E)-2,6-dimethyl-2,4,6-octatriene, 1-nonanol, β-bisabolene, sabinene, 3-carvomenthenone, (4E,6Z)-2,6-dimethyl-2,4,6-octatriene, ipsdienol, and 2-thujene. Lastly, DE variety exclusively contained 2-pentoxyethyl acetate.
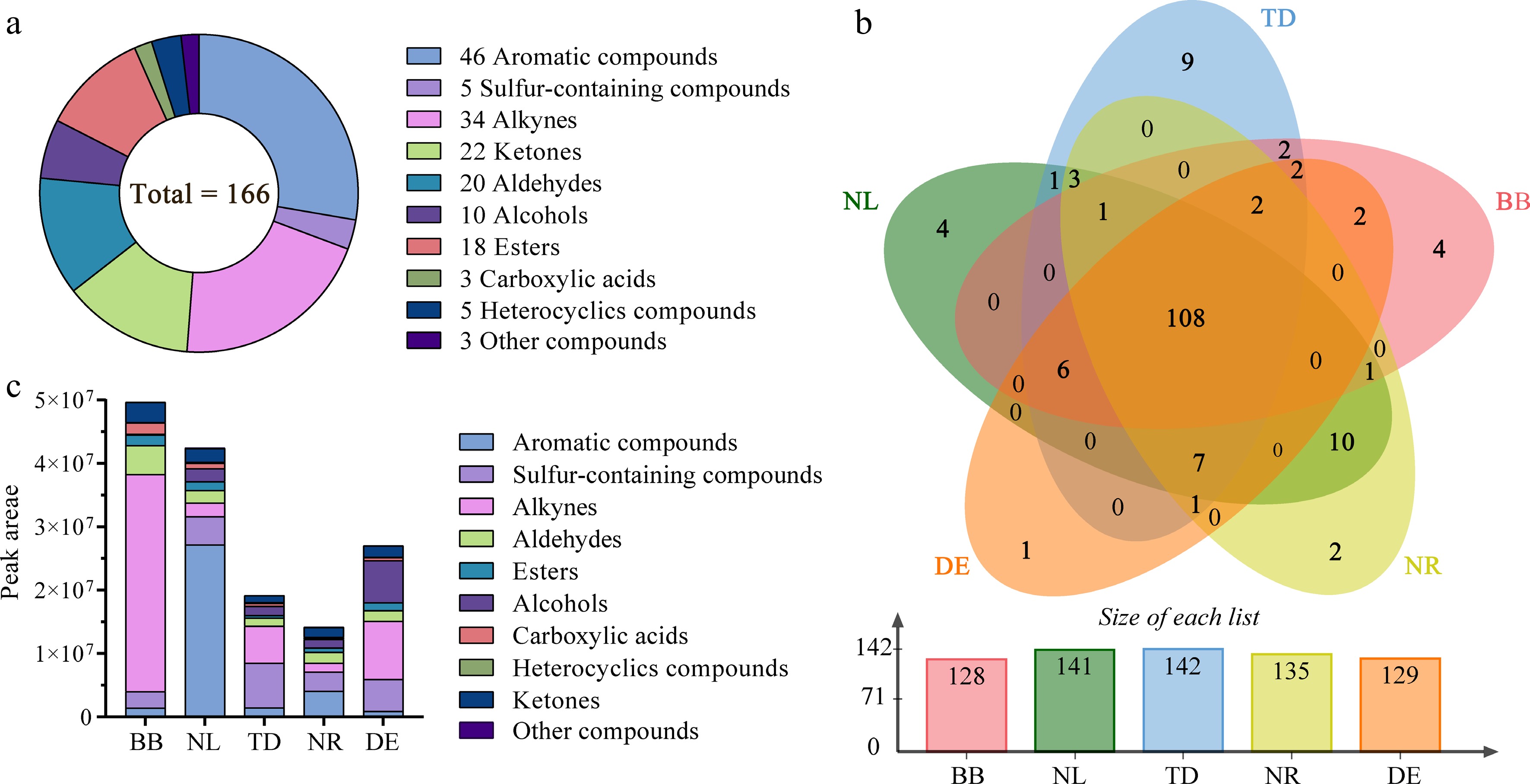
Figure 4.
Overview of the volatile components. (a) Quantitative distribution of chemical classes of volatile compounds. (b) Venn plot; numbers represent the identified metabolites. (c) Relative abundance of different types of volatile compounds. Note: NL, N. lotus; NR, N. rubra; TD, Nymphaea 'Texas Dawn'; BB, Nymphaea 'Blue Bird'; DE, Nymphaea 'Detective Erika'.
The relative content of volatiles calculated from the total ion chromatograms varied in the concentration and proportion of each chemical class in different samples. Among them, the highest peak area of volatile components was found in BB, followed by NL (Fig. 4c). Alkenes were found to be the most abundant volatile components in BB, accounting for 69.58% of the total volatile components. Aromatic compounds were found to be the most abundant volatile components in NL, accounting for 64.25% of the total volatile components. In addition, alcohols and alkenes accounted for 28.05% and 26.89% of the total volatile components in DE, respectively.
We investigated the relative amounts of the main 24 chemical compounds released that were greater than 1% in BB, NL, TD, NR, DE, with total relative contents of 90.92%, 91.45%, 83.19%, 77.29%, and 90.83%, respectively (Table 1 & Fig. 5). The concentrations of the identical chemical compounds varied across distinct samples. Within the provided BB sample, three compounds of the alkene class, namely 1,11-dodecadiene (27.30%), (E)-β-famesene (18.28%), and α-farnesene (14.44%), collectively constitute more than half of the total volatile compounds. Among the NL samples, 2,5-dimethoxytoluene (56.18%) emerged as the most abundant compound. In TD and NR samples, dimethyl sulfide was the predominant volatile compound, comprising 33.61% and 18.75% of the total volatile content, respectively. In addition, in DE sample, the concentration of benzyl alcohol (23.83%) was the most abundant, followed by dimethyl sulfide (16.67%) and α-farnesene (16.41%).
Table 1. Comparison of the main volatile compounds in five water lily samples.
No. Compounds Class RI (ref)[a] RI (cal)[b] CAS Ion Odor type[c] Flavor[c] Relative content (%) BB NL TD NR DE 1 Benzyl alcohol Alcohols 1036 ± 4 1037 100-51-6 79 Floral Floral, rose, balsamic 0.11 4.48 6.32 8.13 23.83 2 Benzaldehyde Aldehydes 962 ± 3 969 100-52-7 77 Fruity Almond, burntsugar, sweet 8.10 2.13 3.17 5.22 3.91 3 Hexanal Aldehydes 800 ± 2 801 66-25-1 41 Green Green, fatty, leafy 0.15 0.69 0.61 3.84 0.34 4 Benzeneacetaldehyde Aldehydes 1045 ± 4 1049 122-78-1 91 Green Green, floral, honey 0.16 0.14 1.03 0.09 0.85 5 1,11-Dodecadiene Alkenes 1179 ± 2 1763 5876-87-9 67 / / 27.30 3.67 12.62 7.04 11.45 6 (E)-β-Famesene Alkenes 1457 ± 2 1453 18794-84-8 41 Woody Woody, citrus, herbal 18.28 0.39 0.84 0.38 1.27 7 α-Farnesene Alkenes 1508 ± 2 1506 502-61-4 41 Woody Citrus, herbal, neroli 14.44 0.17 10.38 0.18 16.41 8 (E)-α-Bergamotene Alkenes 1433 ± 3 1437 13474-59-4 93 Woody Woody 2.02 0.11 0.03 0.03 1.65 9 β-Sesquiphellandrene Alkenes 1524 ± 2 1528 20307-83-9 69 Herbal Herbal, fruity, woody 3.43 0.12 0.15 0.10 1.37 10 (E)-β-Ocimene Alkenes 1049 ± 2 1048 3779-61-1 93 / Herbal, sweet 0.01 0.03 3.89 0.04 0.01 11 2,5-Dimethoxytoluene Aromatic compounds 1251 ± 5 1249 24599-58-4 137 / / 0.38 56.18 0.49 12.18 0.52 12 1,4-Dimethoxybenzene Aromatic compounds 1168 ± 9 1166 150-78-7 123 Green Green, hay, sweet 0.05 3.62 0.02 6.63 0.02 13 Phenol Aromatic compounds 980 ± 4 981 108-95-2 94 Phenolic Phenolic, plastic, rubbery 0.17 0.87 0.56 1.46 0.62 14 Acetic acid Carboxylic acids 610 ± 10 581 64-19-7 45 Acidic Pungent, sour 3.42 2.01 2.18 0.77 1.58 15 Benzoic acid, methyl ester Esters 1094 ± 3 1096 93-58-3 105 Phenolic Phenolic, wintergreen, almond 0.03 0.57 0.07 1.86 0.03 16 Ethyl acetate Esters 612 ± 5 613 141-78-6 43 Ethereal Ethereal fruity sweet 0.38 0.39 0.14 0.00 2.06 17 Acetic acid, phenylmethyl ester Esters 1164 ± 2 1166 140-11-4 108 Floral Floral, fruity, jasmine 1.88 0.01 0.06 0.02 0.45 18 6-Methyl-5-heptene-2-one Ketones 986 ± 2 984 110-93-0 43 Citrus Citrus, green, musty 1.29 2.16 2.15 3.67 3.05 19 (E)-3-Penten-2-one Ketones 735 ± N/A 744 3102-33-8 69 / / 0.21 1.01 0.31 1.58 0.40 20 (E)-Geranylacetone Ketones 1453 ± 2 1448 3796-70-1 43 Floral Fruity, fresh, rose 2.15 0.76 0.65 0.72 1.18 21 2-Heptadecanone Ketones 1902 ± 7 1900 2922-51-2 58 / / 1.68 0.41 1.18 0.70 0.80 22 Dimethyl sulfide Sulfur-containing compounds 520 ± 7 553 75-18-3 47 Sulfurous Sulfurous, sweet corn 4.37 8.76 33.61 18.75 16.67 23 Carbon disulfide Sulfur-containing compounds 549 ± 13 565 75-15-0 76 / Sweet 0.55 1.17 1.96 1.29 1.39 24 Benzothiazole Sulfur-containing compounds 1229 ± 8 1240 95-16-9 135 Sulfurous Sulfurous, rubbery, vegetable, cooked 0.29 0.48 0.56 1.16 0.53 Total relative content (%) 90.92 91.45 83.19 77.29 90.83 RI, Retention index, Ion, Qualitative ion; [a] RI (ref): The RI values (median ± deviation) were the reference values for semi-standard non-polar (DB-5) column obtained from NIST 2014; [b] RI (cal): The RI values were calculated from C8-C40 n-alkanes; [c] odor type and flavor were obtained from website (www.thegoodscentscompany.com/search2.html). Note: NL, N. lotus; NR, N. rubra; TD, N. 'Texas Dawn'; BB, N. 'Blue Bird'; DE, N. 'Detective Erika'. 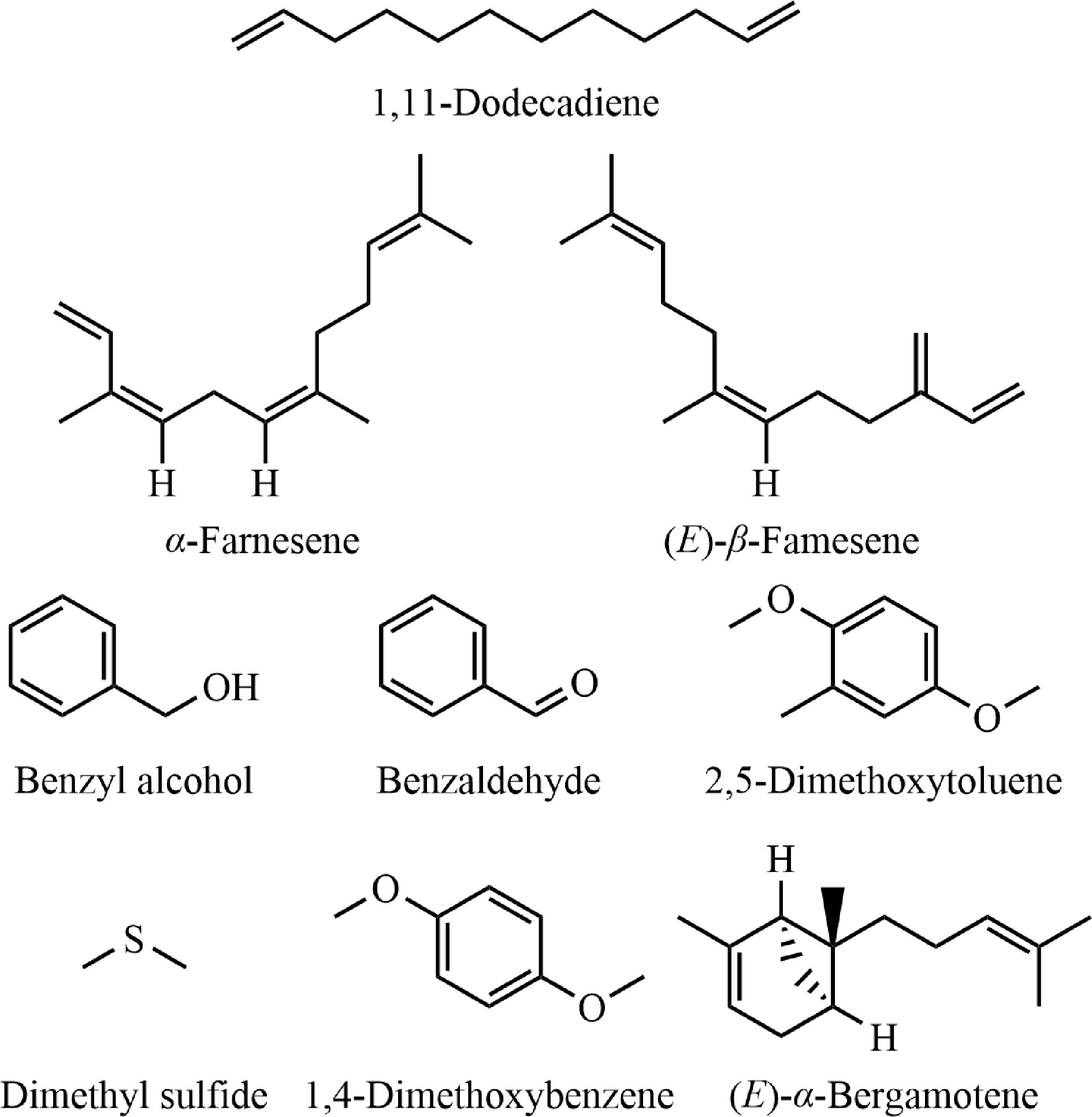
Figure 5.
Molecular formulas of major volatile compounds.
Analysis of different volatiles in water lily samples
-
To gain a comprehensive understanding of the variations in volatile compound content among the five water lily samples, we utilized PLS-DA with the peak areas of 166 volatile compounds as input variables. As shown in Fig. 6a, the five samples were distinctly segregated from the remaining samples along the principal component 1 axis (R2X [1] = 37.6%) and principal component 2 axis (R2X [2] = 27.1%). Cross-validation was performed using the leave-one-out method, with the first two principal components explaining 99.6% of the total variance (R2X). The model exhibited good predictive ability (Q2 = 85.1%) and was not overfitted. The evident segregation and high reproducibility observed among the various sample groups substantiated the presence of significant disparities in the volatile compositions of the five water lily species and varieties (Fig. 6a).
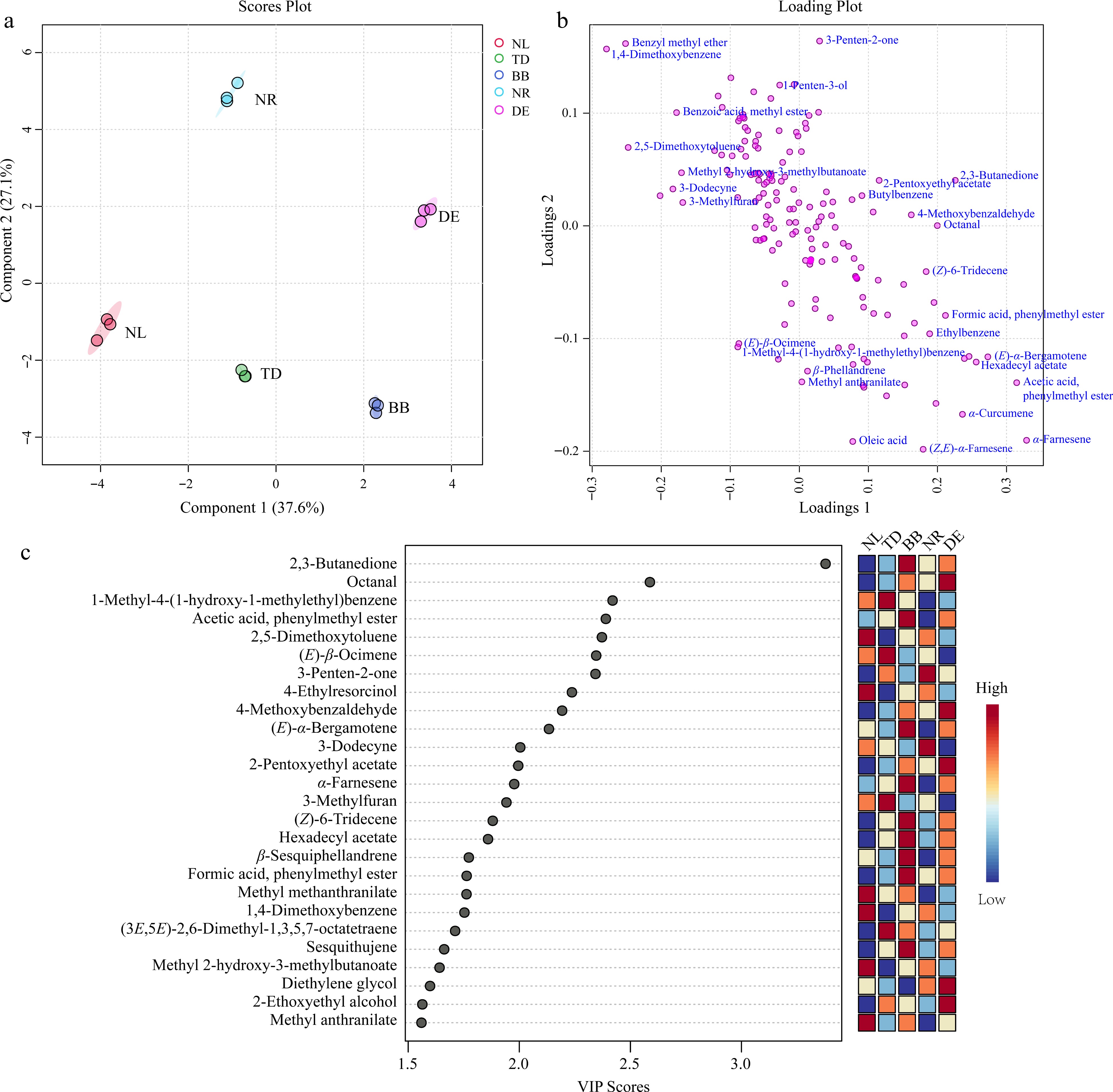
Figure 6.
The partial least squares discriminant analysis (PLS-DA) of the volatile compounds. (a) Score plot of PLS-DA. (b) The loading plot of PLS-DA. (c) Variable importance in the project (VIP) plot of PLS-DA. Note: NL, N. lotus; NR, N. rubra; TD, Nymphaea 'Texas Dawn'; BB, Nymphaea 'Blue Bird'; DE, Nymphaea 'Detective Erika'.
The variable importance in projection (VIP) value is a comprehensive metric that quantifies the contribution of a variable in describing the data and indicates the significance of an independent variable for the model[21]. By utilizing the PLS-DA model, we identified 42 key volatile compounds with VIP scores of ≥ 1 across all samples (Supplemental Fig. S5). Subsequently, a one-way analysis of variance (ANOVA) was conducted on these compounds, revealing statistically significant differences among the distinct sample groups (p < 0.05). Notably, 26 metabolites of volatile compounds exhibited VIP values exceeding 1.5 (Fig. 6c), with 2,3-butanedione, octanal, 1-methyl-4-(1-hydroxy-1-methylethyl)benzene, acetic acid, phenylmethyl ester, 2,5-dimethoxytoluene, (E)-β-ocimene being among the top-ranked compounds in descending order of VIP values.
-
Although previous studies have been reported on the volatile and non-volatile components of water lilies, the number of compounds reported in these studies is relatively limited[2,22]. By employing widely targeted metabolomics, we overcome the limitations associated with both targeted and non-targeted metabolite detection methods. This approach provided us with a high-throughput analysis, increased sensitivity, and wide coverage, enabling a more comprehensive understanding of the metabolomic profile of water lilies. In the present study, we comprehensively identified 533 non-volatile and 166 volatile components in five different colors of water lilies using UPLC-MS/MS and GC × GC-TOFMS techniques.
It is widely recognized that flower color is a crucial characteristic of ornamental plants and is influenced by various factors. Specifically, the type and concentration of anthocyanins are generally considered to be the primary determinants[23]. Anthocyanins play an important role in plant physiology, serving as attractions for pollinators and herbivores, acting as deterrents against herbivores and parasites, and also influencing visual signals and mimicry of defensive structures[24]. Among the diverse range of flower colors, blue coloration primarily results from the presence of anthocyanins derived from delphinidin[25]. In the petals of Nymphaea 'King of Siam', four anthocyanins were identified: delphinidin 3-O-β-galactopyranoside, delphinidin 3′-O-(2″-O-galloyl-β-galactopyranoside, delphinidin 3-O-(6″-O-acetyl-β-glucopyranoside, delphinidin 3′-O-(2″-O-galloyl-6″-O-acetyl-β-galactopyranoside[13]. In the present study, a total of 11 anthocyanins were detected in five different-colored water lilies. Among them, delphinidin-derived anthocyanins exhibited relatively higher relative abundances in BB, TD, and DE, while its abundance was low in DE. Notably, cyanidin-3-O-galactoside, cyanidin-3-O-glucoside, cyanidin-3-rutinoside, cyanidin-O-acetylhexoside, and cyanidin-O-syringic acid, which are glycosides of the cyanidin type, were found to be the most abundant exclusively in the DE samples. In contrast, these compounds were found at very low levels or were not detected in the other four water lily samples.
In addition, numerous studies conducted on various plant species have provided evidence that flavonoids (including flavones, flavanols, and isoflavonoids) and their glycosides play a significant role in co-pigmentation[26,27]. According to reports, N. lotus stamen extracts contain a higher concentration of flavonoids compared to perianth extracts[22]. The flavonoids identified in the stamen extracts include kaempferol 3-O-galactoside, quercetin 3′-O-xyloside, quercetin 3-O-rhamnoside, isorhamnetin 7-O-galactoside, and myricetin 3′-O-xyloside. Another study indicated that the content of flavonoids in the petals of the N. alba flower is significantly higher than in the stem and root[28]. In addition, a large number of phenolic acids, for example, caffeic acid, chlorogenic acid, p-coumaric acid have been identified in water lilies[12]. Our present study identified various flavonoid glycosides with glucose, rhamnose, galactose, and arabinose as the major sugar ligands. As a result, the number of detected flavonoids and their glycosides in water lilies has significantly expanded. Among them, quercetin, kaempferol, apigenin, myricetin, and luteolin were identified as the five major flavonols present in water lilies. In addition, many previous studies have reported the antioxidant potential of water lily extracts that are associated with the accumulation of its phytochemicals, especially flavonoids[5,7,29]. Therefore, the variations in the composition of flavonoids among distinct water lily species and varieties, as revealed in this investigation, are expected to provide valuable insights into their diverse antioxidant properties.
Water lilies not only exhibit a wide variety of colors throughout their flowering period but also emit a captivating fragrance, which has garnered significant attention among researchers in recent studies[2,30]. Among the seven species of N. subg. Hydrocallis (Nymphaeaceae), N. lotus was only found to contain detectable levels of 2,5-dimethoxytoluene, and the content was notably high[16]. This is consistent with the results of our study. Dimethyl sulfide, a group of sulfur-containing compounds, was detected at significant concentrations in all five water lilies. Notably, it has been identified as a crucial aromatic volatile in green tea[31] and mandarin juices[32], and even in trace amounts, it contributes to the development of sulfurous notes. Benzyl alcohol, renowned for its delightful floral and sweet essence, has been extensively documented in various Nymphaea species[2]. Its relative concentration in the DE samples was 23.83% of the total volatiles. This high concentration may be the main reason for the sweet and floral aroma of the DE samples. Additionally, according to the report, 2,3-butanedione is known for its buttery, sweet, and creamy flavor, which imparts a cheese-like taste to soy milk[33]. Our research findings indicate that the BB samples had the highest concentration of 2,3-butanedione, which may be responsible for its sweet and creamy flavor, while it was not detected in the NL sample.
In addition, previous studies reported that the flower aroma of the cold-resistant water lily is influenced by nerolidol and lilac alcohols, with orange and clove flower aromas, while the aroma of tropical water lilies is influenced by ethyl benzoate, acetic acid phenylmethyl ester, and 2-heptadecanone, which provide a fruity and sweet flower aroma[2]. Moreover, the primary volatile compounds identified in the flowers of Nymphaea hybrida were benzyl alcohol, benzyl acetate, benzaldehyde, (E)-α-bergamotene, and these may constitute its main fragrant components[34]. We observed that the NL sample releases a greater proportion of aromatic compounds than the other samples, which may account for its fruity and sweet flavor. In addition, (E)-α-Bergamotene, (E)-β-famesene and 11 other different volatile compounds were reported in N. colorata flowers, which may serve as olfactory cues for insect pollinators[35]. (E)-β-famesene and α-farnesene possess woody, citrus, and herbal aromatic attributes, was found to be more pronounced in BB samples, accounting for 18.28% and 14.44% of the total volatile composition, respectively. These compounds are likely responsible for the woody fragrance observed in BB samples. Overall, the varying concentrations and relative amounts of volatile compounds contribute to the rich and diverse fragrance qualities of these five water lily species and varieties.
-
To sum up, our study utilized advanced widely targeted metabolomics to comprehensively investigate the non-volatile and volatile components in five water lily species and varieties. The results revealed significant differences in the composition and abundance of metabolites among the different colors of water lily samples. Regarding non-volatile components, we found that cyanidin-type anthocyanins were abundant in the purple-colored DE samples, while delphinidin-derived anthocyanins were prominent in BB, which exhibited a blue color. Flavonoids, phenolic acids, amino acids, lipids, and organic acids were found to be the dominant non-volatile metabolites, expanding our knowledge of their metabolic profile. Pathway enrichment analysis of the differential compounds indicated the involvement of various metabolic pathways such as flavonoid biosynthesis and anthocyanin biosynthesis in water lily metabolism. In terms of volatile components, water lilies contain diverse volatile compounds, including aromatic compounds, alkynes, ketones, alcohols, esters, and aldehydes. Among these, the concentration ofkey compounds including 1,11-dodecadiene, benzyl alcohol, benzaldehyde, α-farnesene and dimethyl sulfide, showed significant differences among samples. By investigating both non-volatile and volatile metabolites, we have obtained a comprehensive understanding of the metabolic pathways and interplay within the water lily species and varieties. Nevertheless, the intricate mechanisms underlying the synthesis and release of metabolites in water lilies warrant further investigation.
-
The authors confirm contribution to the paper as follows: study conception and design: Chen Y, Lv H; data collection: Wei J, Wu Y; resources: Chen S, Yu C; analysis and interpretation of results: Lin Z, Zhu Y; draft manuscript preparation: Yang G. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are not publicly available due to management requests, but are available from the corresponding author on reasonable request.
This work was supported by the Science and Technology Innovation Project of Chinese Academy of Agricultural Sciences (CAAS-ASTIP-2014-TRICAAS) for financial support.
-
The authors declare that they have no conflict of interest.
- Supplemental Table S1 Crucial changed differential non-volatile metabolites in five water lily samples.
- Supplemental Fig. S1 The spectrogram of non-volatile components in five water lily samples obtained from UPLC-MS/MS.
- Supplemental Fig. S2 Heatmap of crucially changed differential non-volatile metabolites in five water lily samples.
- Supplemental Fig. S3 Differential non-volatile metabolite KEGG enrichment maps.
- Supplemental Fig. S4 The spectrogram of volatile components in five water lily samples obtained from GC×GC-TOFMS.
- Supplemental Fig. S5 The heat map of volatile compounds with VIP scores of ≥1.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Yang G, Wei J, Wu Y, Chen S, Yu C, et al. 2024. Comprehensive study of non-volatile and volatile metabolites in five water lily species and varieties (Nymphaea spp.) using widely targeted metabolomics. Beverage Plant Research 4: e012 doi: 10.48130/bpr-0024-0005 

Comprehensive study of non-volatile and volatile metabolites in five water lily species and varieties (Nymphaea spp.) using widely targeted metabolomics
- Received: 09 November 2023
- Revised: 21 December 2023
- Accepted: 10 January 2024
- Published online: 03 April 2024
Abstract: Water lilies, members of the Nymphaeaceae family, are globally cultivated aquatic plants known for their diverse colors and significant ornamental, economic, beverage, medicinal, and ecological value. In this study, we employed ultra-high performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) to analyze the non-volatile components and simultaneous distillation extraction (SDE) in combination with two-dimensional gas chromatography-time-of-flight mass spectrometry (GC×GC-TOFMS) to analyze the volatile components in five water lily species and varieties. Results showed that 118 differential metabolites including flavonoids, phenolic acids, amino acids, and lipids were screened among 533 non-volatiles. Cyanidin-type anthocyanins, including cyanidin-3-O-galactoside, cyanidin-3-O-glucoside, and cyanidin-3-rutinoside, are present in high amounts in the purple-colored Nymphaea 'Detective Erika'. Conversely, delphinidin is found in significant quantities in Nymphaea 'Blue Bird', which exhibits a blue color. KEGG analysis showed that flavonoid biosynthesis and anthocyanin biosynthesis exhibited significant enrichment. Additionally, a total of 166 volatiles were screened in water lilies, mainly including aromatic compounds, alkynes, ketones, alcohols and esters. Among them, the concentrations of key compounds including 1,11-dodecadiene, benzyl alcohol, benzaldehyde, α-farnesene and dimethyl sulfide, varied significantly among different samples. This study reveals significant variations in chemical compounds among different Nymphaea species and varieties. These findings contribute to enhancing our comprehension of the metabolic variability and composition of water lilies, which might shed light on unlocking new possibilities for their potential application in the beverage industry.