









-
Melon (Cucumis Melo L.), a genus of the Cucurbitaceae family, is an important cash crop cultivated worldwide[1]. It is widely grown on the world's five continents. According to FAO data (
www.fao.org ), global melon production in 2022 was 28.56 million tons, with China accounting for about half of this production. In China, Xinjiang is one of the main producing areas, consistently ranking first in melon yield throughout the year. Cantaloupe belongs to the calabash family, melon genus, and thick-skinned melon class, and is a traditional specialty of Xinjiang, China. It has a life cycle of 80–120 d, and it generally takes around 35–45 d to mature after flowering.Flavor (aroma and taste) and nutritional composition are key factors that determine the quality of melon. The rich nutritional value and distinctive flavor of the fruits are often influenced by essential metabolites such as soluble sugars (sucrose and fructose) and organic acids (citric acid, malic acid, etc.), which contribute to the flavor of the fruit[2]. In addition, fruits contain active substances like amino acids and antioxidant vitamins, which play an important role in maintaining human health and slowing down the aging process[3]. Therefore, enhancing fruit flavor and increasing the content of functional active substances not only makes fruits more appealing to consumers but also brings significant benefits to the human health industry. However, improving fruit flavor is not a simple experimental process; it requires a comprehensive understanding of the metabolic pathways and potential metabolic regulatory networks governing the desired fruit flavor quality.
With the development of omics technology, research on fruit metabolic pathways has expanded significantly. Omics technology has led to the discovery of a greater number of genes involved in regulating metabolism. For example, Zhao et al. used metabonomics and transcriptomic analysis to study the metabolic network during the development and maturation of cashew fruits, uncovering potential regulatory factors of phosphatidylinositol biosynthesis[4]. In another study, Hou et al. conducted transcriptome analysis on different varieties of jujube, identifying highly expressed genes associated with jujube cracking. This research provided novel genetic resources for understanding the mechanism behind fruit cracking[5]. Sucrose, being an important sugar in fruits, has also been extensively studied in terms of its metabolic genes[6, 7].
Melon pulp possesses a distinct juicy flavor that is highly favored by consumers worldwide. It is abundant in various metabolites, including amino acids, sugars, organic acids, terpenoid peptides, and analogues[8]. In recent years, extensive research has been conducted on the regulatory mechanisms underlying melon fruit metabolism. For instance, transcriptome analysis has shed lights on the changes in fresh-cut melon fruit quality during storage[9]. High-throughput sequencing has been employed to analyze stem and soil samples of reticulated melon and Oriental melon, providing new insights into reticulate formation in reticulated melon[10]. To improve the quality of melon fruit, ten different pumpkin rootstocks were grafted for melon cultivation. Through metabolite and sensory analysis, the melon variety with the best overall quality was identified[11]. Zhao et al. studied the transcription factor CmNAC34, which regulates the CmLCyb-mediated beta-carotene accumulation during melon fruit ripening, revealing the metabolic regulatory mechanism of carotenoids during melon fruit ripening[12]. However, it is important to note that most of these studies were conducted under specific developmental conditions and focused on post-ripening fruits. Utilizing only transcriptomics or metabolomics may not provide a comprehensive understanding of the complete metabolic changes occurring throughout the entire developmental stage of melon fruits.
As a result, we used the 'Xizhou Mi 25' melon, the most common variety growing in China's Xinjiang area, as our primary research object to investigate the physical properties of fruit at various developmental phases. Transcriptomics and metabolomics were utilized to investigate changes in key genes and metabolites at different phases of fruit growth. The molecular mechanism of important quality metabolism in melon development was explored using gene functional analysis, metabolite function analysis, and combined metabolite-gene analysis. Our findings may be used to improve and adapt melon taste quality, solving industrial development problems.
-
The melon 'Xizhoumi 25' was used as the test material and planted in the comprehensive experimental field of the Xinjiang Academy of Agricultural Sciences in 2022. Samples were collected at 5, 15, 25, 35, and 45 d after flowering. For each stage, three biological replicates were prepared, with each replicate consisting of ten melons. in addition to sample collection, various parameters such as horizontal and vertical diameter, soluble solids, weight, hardness, and other indicators were measured. The collected samples were quickly frozen with liquid nitrogen and ground into a powder with a grinder, then stored in a refrigerator at −80 °C for future use. The samples were designated as M5D to M45D for subsequent analyses.
Metabolomic analysis
Sample preparation
-
Fifty mg tissues were weighed and extracted by directly adding 800 μL of precooled extraction reagent (MeOH : H2O, 70:30, v/v, precooled at −20 °C). To ensure sample preparation quality control, 20 μL of an internal standards mix was added (0.02 mg/mL L-2-chlorophenylalanine). Two small steel balls were placed in the Eppendorf tube, and homogenization was performed at 50 Hz for 5 min using TissueLyser (JXFSTPRP, China). The samples were then sonicated for 30 min at 4 °C and incubated at −20 °C for 1 h. Following this, the samples were further centrifuged at 14,000 rpm, 4 °C, for 15 min. Subsequently, 600 μL of the supernatant was filtered through 0.22 μm microfilters and transferred to autosampler vials for LC-MS analysis. A quality control (QC) sample was prepared by pooling 20 μL of the supernatant from each sample to evaluate the reproducibility and stability of the entire LC-MS analysis.
UHPLC-MS analysis
-
Sample analysis was performed using a Waters ACQUITY UPLC 2D system (Waters, USA), coupled with a Q-Exactive mass spectrometer (Thermo Fisher Scientific, USA) with a heated electrospray ionization (HESI) source. Chromatographic separation was achieved using a Hypersil GOLD aQ column (2.1 mm × 100 mm, 1.9 μm, Thermo Fisher Scientific, USA), where mobile phase A was composed of 0.1% formic acid in water and mobile phase B was composed of 0.1 formic acid in acetonitrile. The column temperature was maintained at 40 °C. The gradient conditions were as follows: 5% B from 0.0 to 2.0 min, 5%−95% B over 2.0−22.0 min, holding at 95% B from 22.0 to 27.0 min, and a wash step with 95% B over 27.1−30 min. The flow rate was set at 0.3 mL/min, and the injection volume was 5 μL.
The mass spectrometric settings for positive and negative ionization modes were configured as follows: for positive ionization mode, the spray voltage was set to 3.8 kV, while for negative ionization mode, it was set to −3.2 kV. The sheath gas flow rate was maintained at 40 arbitrary units (arb), and the auxiliary (aux) gas flow rate was set to 10 arb. The aux gas heater temperature was set to 350 °C, and the capillary temperature was set to 320 °C. In full scan mode, the mass spectrometer scanned the range of 100–1,500 m/z with a resolution of 70,000. The automatic gain control (AGC) target for MS acquisitions was set to 1e6, and the maximum ion injection time was 100 ms. For MS/MS fragmentation, the top three precursors were selected, and the maximum ion injection time was set to 50 ms with a resolution of 30,000. The AGC target for MS/MS acquisitions was set to 2e5. The stepped normalized collision energy was applied at 20, 40, and 60 eV for fragmentation.
Data preprocessing and annotation
-
After the mass spectrometry detection is completed, the raw data of LC/MS is preprocessed by Progenesis QI (Waters Corporation, Milford, USA) software, and a three-dimensional data matrix in CSV format is exported. The information in this three-dimensional matrix includes sample information, metabolite name, and mass spectral response intensity. Internal standard peaks, as well as any known false positive peaks (including noise, column bleed, and derivatized reagent peaks), were removed from the data matrix, deredundant, and peak pooled. At the same time, the metabolites were searched and identified, and the main database was the HMDB (
www.hmdb.ca ), Metlin (https://metlin.scripps.edu) and Majorbio Database.Differential metabolites analysis
-
Perform variance analysis on the matrix file after data preprocessing. The R package ropls (Version 1.6.2) performed principal component analysis (PCA) and orthogonal least partial squares discriminant analysis (OPLS-DA), and used 7-cycle interactive validation to evaluate the stability of the model. In addition, student's t-test, and fold difference analysis were performed. The selection of significantly different metabolites was determined based on the variable importance in the projection (VIP) obtained by the OPLS-DA model and the p-value of student's t-test, and the metabolites with VIP > 1, p < 0.05 were significantly different metabolites.
RNA-seq analysis
RNA extraction
-
Total RNA was extracted from the tissue using TRIzol® Reagent, specifically the Plant RNA Purification Reagent designed for plant tissue, according to the manufacturer's instructions. Subsequently, the quality of the RNA was assessed using the 5300 Bioanalyser from Agilent, and the concentration was determined using the ND-2000 (NanoDrop Technologies). Only RNA sample with high quality, meeting the following criteria, was used for the construction of the sequencing library: OD260/280 ranging from 1.8 to 2.2, OD260/230 greater than or equal to 2.0, RIN equal to or greater than 6.5, 28S:18S greater than or equal to 1.0, and a quantity of more than 1 μg.
Library preparation and sequencing
-
RNA purification, reverse transcription, library construction, and sequencing were performed at Shanghai Majorbio Bio-pharm Biotechnology Co., Ltd. (Shanghai, China), according to the manufacturer's instructions (Illumina, San Diego, CA, USA). The melon RNA-seq transcriptome library was prepared following Illumina® Stranded mRNA Prep, Ligation kit. A total of 1μg of total RNA was used for the library construction process. The procedure involved the isolation of messenger RNA using oligo(dT) beads for polyA selection. The isolated mRNA was then fragmented using a fragmentation buffer. Subsequently, double-stranded cDNA was synthesized using the SuperScript double-stranded cDNA synthesis kit (Invitrogen, CA, USA) with random hexamer primers from Illumina. The synthesized cDNA underwent end-repair, phosphorylation, and 'A' base addition, according to Illumina's library construction protocol. The libraries were size-selected for cDNA target fragments of 300 bp using 2% Low Range Ultra Agarose, followed by PCR amplification using Phusion DNA polymerase (NEB) for 15 PCR cycles. The libraries were quantified using the Qubit 4.0 instrument, and paired-end RNA-seq sequencing was performed using the NovaSeq 6000 sequencer with a read length of 2 × 150 bp.
Quality control and read mapping
-
The raw paired-end reads were subjected to trimming and quality control using FASTP[13] with default parameters. Then the clean reads were separately aligned to a reference genome in orientation mode using HISAT2 software[14]. The mapped reads of each sample were assembled using StringTie software[15] in a reference-based approach.
Differential expression analysis and functional enrichment
-
To identify DEGs (differential expression genes) between two different samples, the expression level of each transcript was calculated using the transcripts per million reads (TPM) method. RSEM[16] was used to quantify gene abundances. Differential expression analysis was performed using either DESeq2[17] or DEGseq[18]. DEGs with |log2FC| ≥ 1 and FDR ≤ 0.05(DESeq2) or FDR ≤ 0.001(DEGseq) were considered significantly differentially expressed. In addition, functional enrichment analysis, including GO and KEGG analysis, were performed to identify DEGs that were significantly enriched in GO terms and metabolic pathways. The analysis was conducted at Bonferroni-corrected p-value ≤ 0.05, compared to the whole-transcriptome background. GO functional enrichment and KEGG pathway analysis were carried out using Goatools and KOBAS[19], respectively.
Data processing and statistical analysis
-
The data were subjected to a one-way analysis of variance, and mean separations were compared using Duncan's multiple mutation tests[20,21]. The results were presented as mean ± standard error. P-value less than 0.05 (p < 0.05) was considered statistically significant. Co-expression and hierarchical clustering (HCL) analyses were performed using the free online platform of Majorbio Choud (cloud.majorbio.com). These analyses allow for the identification of genes or samples that exhibit similar expression patterns and their grouping based on similarity.
-
The phenotypic changes of melon at different stages of development are shown in Fig. 1. At 25 d after flowering, a slight reticulation started to emerge, accompanied by an emerald green skin color. By 35 d, a significant amount of mesh patterning appeared, and the color changed from emerald green to light green. At 45 d, the melon's surface was covered by a large amount of white mesh.

Figure 1.
Appearance changes of melon at different days post anthesis.
Analysis of the longitudinal and transverse diameters showed that before 35 d, the longitudinal diameter was smaller than the transverse diameter. However, at 45 d, the longitudinal diameter exceeded the transverse diameter, indicating that the melon elongates more than it expands width-wise between 35 and 45 d, resulting in an oval shape. The melon's weight underwent the most significant changes at 5~15 d and 15~25 d. At 35 d, the melon's weight became similar to that at 45 d. At maturity, the average melon weight was 2,170.08 ± 142.17 g. Regarding hardness, it was highest at 15 d, significantly differing from other stages. As the fruit ripens, the hardness gradually decreases. Soluble solids, an important indicator for assessing muskmelon quality, showed an overall increasing trend from 5 to 45 d development, and at 45 d, the soluble solids content exceeded 15% (Table 1).
Table 1. Fruit phenotypes of muskmelon at different developmental stages.
Days after flowering (day) Weight
(g)Longitudinal (cm)/transverse diameter (cm) Hardness (kg/m3) Soluble solid (%) 5 144 ± 11.97d / 5.01 ± 1.58c 6.35 ± 0.52e 15 837.42 ± 81.43c 0.72 ± 0.04c 6.13 ± 0.17a 8.14 ± 0.2c 25 1523.24 ± 97.22b 0.75 ± 0.02b 5.58 ± 1.42b 6.79 ± 0.59d 35 2045.58 ± 153.45a 0.76 ± 0.05b 4.98 ± 0.38c 10.84 ± 0.43b 45 2170.08 ± 142.17a 1.07 ± 0.05a 4.41 ± 0.001d 15.16 ± 0.51a Values in the same line with different letters indicate statistically significant differences at p < 0.05. Nontargeted metabonomics during melon fruit development
-
To investigate the dynamics of metabolites during melon fruit development and ripening, a metabolomic identification at five stages (from 5 d after flowering to 45 d) were employed. PCA revealed distinct differences in metabolite compounds at different stages (Fig. 2a). The compounds at 5 and 15 d compounds are similar, as are the compounds at 25 and 35 d, while the metabolic compounds at 45 d differ greatly from other developmental stages. Hierarchical clustering analysis also showed consistent clustering patterns with PCA.
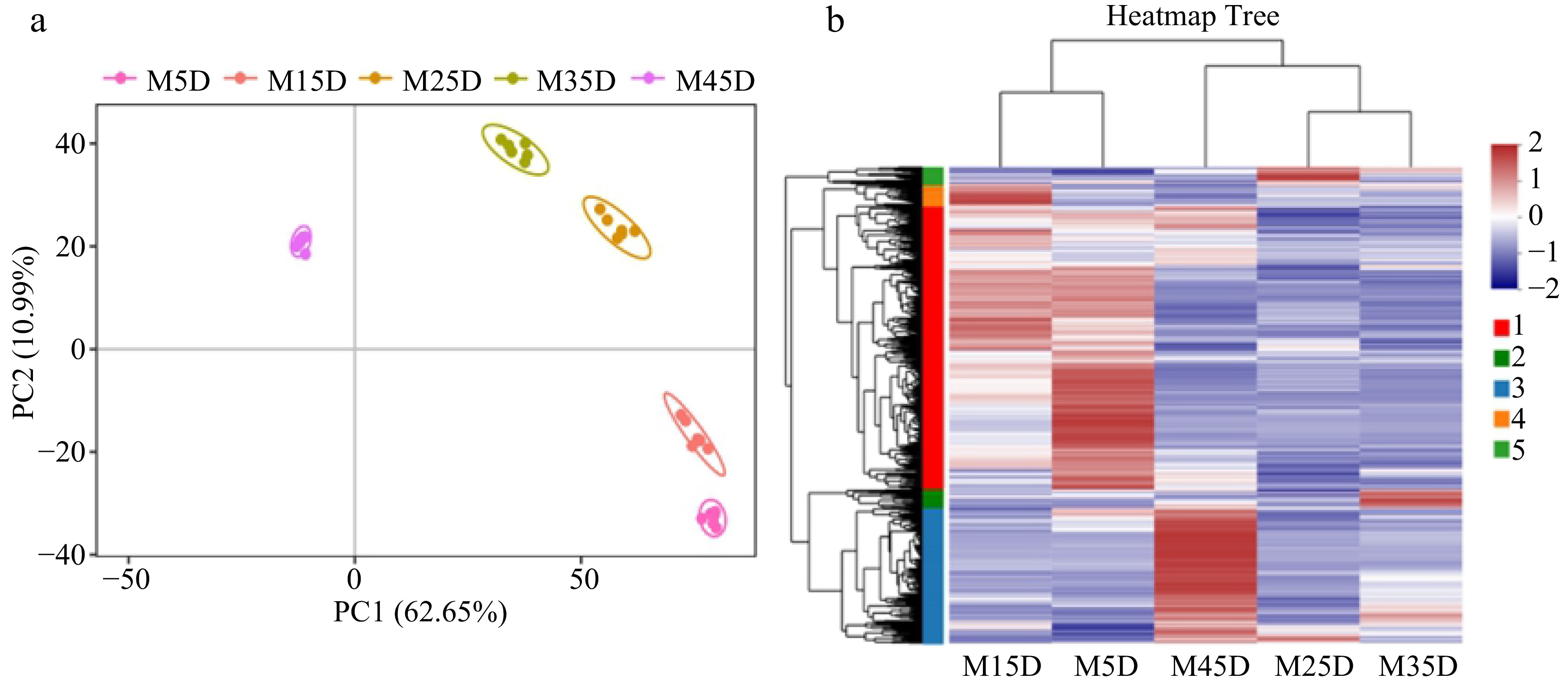
Figure 2.
Metabolome analysis of five developmental stages in melon fruit. (a) PCA of metabolome data. (b) Heat map of metabolites from five developmental stages.
In total, 666 annotated metabolites (431 in positive ion modes and 235 in negative ion modes) were detected by UHPLC-QTOF-MS (Supplemental Table S1). The overall change in total metabolite content showed significant variations in melon fruit metabolites at the five developmental stages (Fig. 2b). Notably, the major differences in metabolites were manifested in the early stage (5 and 15 d) and the mature stage (45 d) of melon development. At 5 d, the compounds with high content mainly included alkaloids, amino acids, and benzene derivatives, indicating biochemical accumulation during early melon development. At 45 d, carbohydrates, alcohols, lipids, and other substances related to the formation of flavor were predominantly found.
To identify significant differences in metabolites during melon development and ripening, VIP ≥ 1.0 and fold change ≥ 1.2 or ≤ 0.83, with a p-value < 0.05 as threshold were set[4]. From the 666 metabolites noted, a total of 268 differential metabolites were obtained (Table 2). In the comparison of M5D vs M15D, 162 differential metabolites were identified (59 upregulated and 103 downregulated). Similarly, M15D vs M25D, M25D vs M35D, and M35D vs M45D comparisons yielded 174, 180, and 180 differential metabolites, respectively (Table 2).
Table 2. Statistics of differentially regulated metabolites 271.
Group M5D:M15D M15D:M25D M25D:M35D M35D:M45D Total Up 59 79 89 91 268 Down 103 95 91 89 All 162 174 180 180 For functional annotation, the KEGG database was used and it was found that the majority of metabolites were identified in the global and overview maps. Amino acid metabolism, biosynthesis of secondary metabolites, carbohydrate metabolism, and lipid metabolism were also significant pathways. KEGG enrichment and concentration analysis showed that 174 metabolites were annotated at different development stages, with the biosynthesis of secondary metabolites exhibiting the most enrichment differences. Additionally, the citrate cycle (TCA cycle), biosynthesis of amino acids, phenylpropanoid biosynthesis, and phenylalanine metabolism were enriched with a greater number of differential metabolites (Supplemental Table S2).
Transcriptomic analysis during fruit development
-
To explore the dynamics of transcriptomes during melon development, 15 libraries consisting of five samples with three replicates per sample were sequenced. On average, 40.06 million raw reads were obtained. After quality control using FASTP software, the average number of clean reads per sample was 44.78 million, with Q30% scores greater than 94.48%. These clean reads were then aligned to the reference genome, resulting in an average alignment rate of 83.39% across the 15 samples, indicating the successful mapping of most of the data to the reference genome (Supplemental Table S3).
Expression levels were quantified in transcripts per million (TPM) using uniquely mapped reads. Genes with TPM > 0 were considered expressed genes. Differentially expressed genes (DEGs) were identified based on meeting the criteria of p-adjust < 0.05 and |log2FC| ≥ 1. A total of 15,487 genes were found to be expressed in melon, of which 10,947 were differentially expressed. Among the four comparison groups (M5D vs M15D, M15D vs M25D, M25D vs M35D, and M35D vs M45D), there were 2,926, 3,691, 3,400, and 2,882 DEGs, respectively. The number of downregulated genes was larger than the number of upregulated genes in each group (Table 3).
Table 3. Statistical table of different genes in different groups.
Compared samples Total no. of DEGs with significant difference Total no. of DEGs significantly up-regulated Total no. of DEGs significantly down-regulated M5D_vs_M15D 2926 924 2002 M15D_vs_M25D 3691 1308 2383 M25D_vs_M35D 3400 1662 1738 M35D_vs_M45D 2882 896 1986 To gain further insights into the 10,947 DEGs obtained, GO annotation classification and KEGG enrichment analysis were performed. In the cell component subcategory, terms such as 'membrane part' and 'cell part' were the most significantly enriched. The molecular function subcategory showed an abundance of terms related to 'binding' and 'catalytic activity'. In the biological processes category, 'metabolic process' and 'cellular process' were the most enriched terms (Fig. 3).
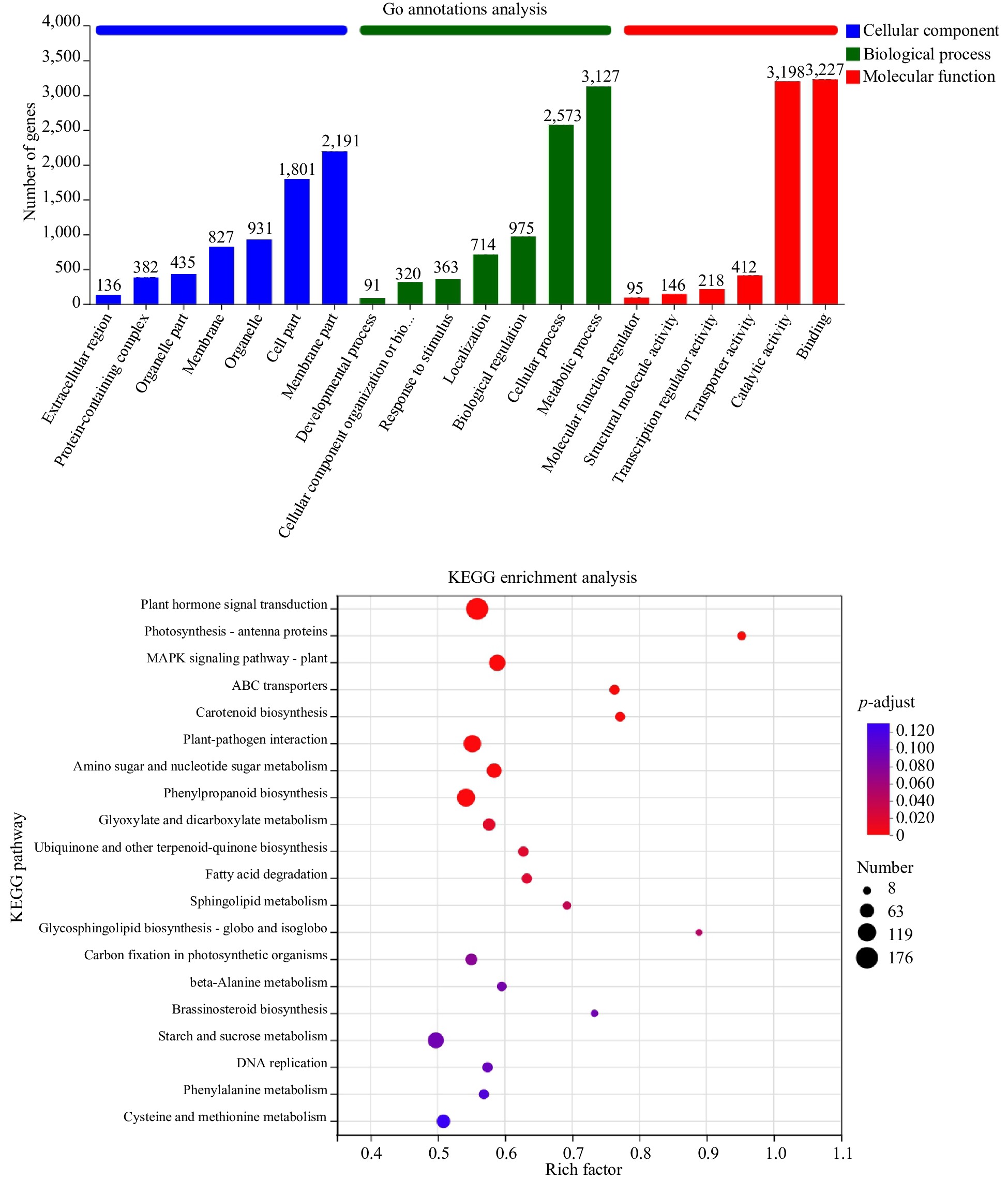
Figure 3.
GO annotation classification and KEGG enrichment analysis of differential genes.
KEGG analysis revealed significant enrichment (p < 0.05) of DEGs in 13 metabolic pathways. The pathway with the largest number of enriched DEGs was 'plant hormone signal transduction', followed by 'phenylpropanoid biosynthesis' and 'plant-pathogen interaction' (Fig. 3). Additionally, pathways such as 'fatty acid degradation' (related to fatty acid metabolism) and 'starch and sucrose metabolism' (key pathway for sucrose accumulation during melon ripening) were also enriched with a considerable number of DEGs[22].
Metabolome and transcriptome are coregulated in clusters that correspond to developmental stages
-
To elucidate the metabolic changes occurring during melon fruit growth, the k-means clustering technique was employed to categorize the 666 metabolites into five clusters (Figs 2b, 4). Cluster 1 contained the largest number of metabolites, which exhibited a gradual decline throughout the five developmental phases. Cluster 4 comprised 29 metabolites that accumulated at high levels during early fruit development. Metabolites in Clusters 2 and 5 accumulate significantly during the middle stage of melon fruit development. Cluster 3 exhibited a gradual accumulation of metabolites, with a total of 189 metabolites enriched metabolites that showed a substantial increase in the later stages of development, closely associated with fruit ripening.

Figure 4.
Transcriptome analysis and k-means cluster analysis of metabolites and genes.
To identify the correlation between gene expression patterns and metabolite accumulation, a co-expression analysis was performed using the metabolite and transcriptome data. Multiple test correction (r > 0.9) was used to check for correlations between genes and metabolites. A total of 9,377 genes were identified to be co-regulated with at least one metabolite, and these genes were further divided into five co-expression clusters (Fig. 4; Supplemental Fig. S1). Remarkably, the five gene clusters displayed similarities to the five metabolite clusters. By studying the correlation between these metabolites and genes, it becomes possible to study and identify some metabolic regulatory networks in melon fruit.
Identification of the genes related to the regulation of soluble sugar accumulation and metabolism
-
In melon fruit, the metabolism and accumulation of soluble sugars play a crucial role in quality formation, particularly during the ripening process. The sweetness of fruit, derived from metabolism processes, is one of the most important quality characteristics and a key factor in attracting consumers. Soluble sugar in melon, such as fructose, sucrose, and maltose, contributes to its sweetness. Sucrose accumulation is particularly important during the late stages of melon fruit development and is a key metabolic pathway for the formation of fruit flavor[23]. In this study, sucrose accumulation was found to be primarily associated with the metabolic pathways of fructose and starch.
In the present analysis of genes related to sugar metabolism, 15 genes that are significantly associated with glucose metabolism were identified, as determined through gene clustering and KEGG annotation analysis (Fig. 5b). These genes are involved in the synthesis and breakdown of soluble sugars such as sucrose. Subcluster 1 consists of five genes, including three HK genes, one SPP gene, and one UGP2 gene. Subcluster 3 includes two genes related to SUS and two genes related to INV. Subcluster 4 contains three genes related to PYG and three genes related to SPS.
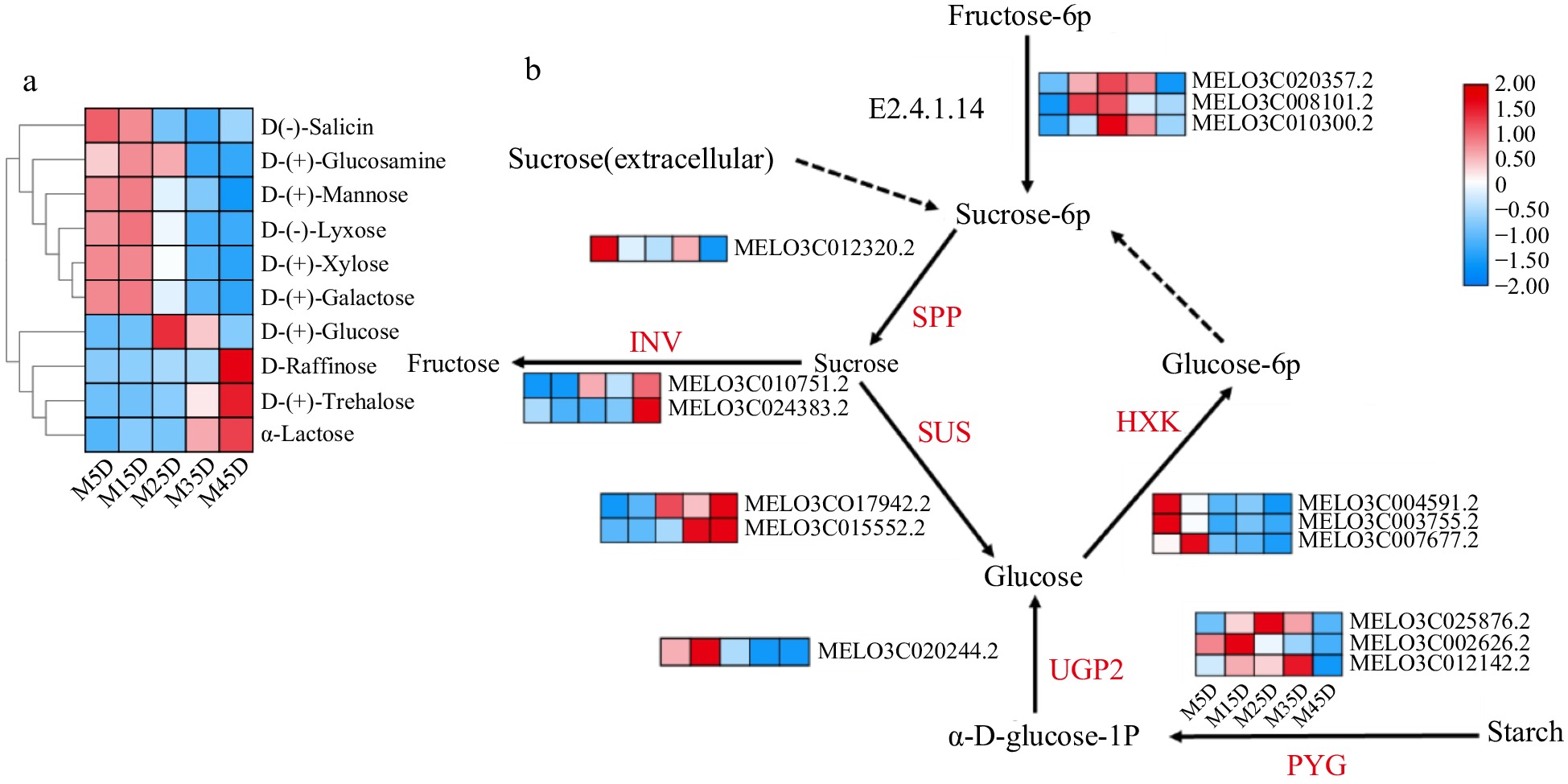
Figure 5.
The expression pattern of genes involved in soluble sugar accumulation in melon at five developmental stages. The development progression of gene expression from M5D to M45D is indicated in five box strings. SPP, sucrose-phosphatase; SUS, sucrose synthase; INV, invertase; HXK, hexokinase; PYG, glycogen phosphorylase; UGP2, UTP-glucose-1-phosphate uridylyltransferase; E2.4.1.14: SPS, Sucrose-phosphate synthase.
SUS is a well-known key enzyme involved in sucrose synthesis. We found that the expression level of SUS was highest in the late development stage (M45D), which is consistent with the accumulation of sucrose during the later stages of melon fruit development. Starch degradation, another marker of fruit ripening was indicated by a significant decrease in the expression level of starch-related enzymes, particularly PYG, in the later stages of melon development.
Furthermore, by analyzing the metabolomic data, 10 metabolites associated with sugar metabolism were identified. These metabolites include D-Raffinose, D-Glucosamine, α-Lactose, D-Trehalose, D-Salicin, D-Lyxose, D-Xylose, D-Galactose, D-Glucose, and D-Mannose (Fig. 5a). These metabolites are involved in various pathways related to sugar metabolism and provide further insights into the synthesis pathway of soluble sugars, including sucrose, in melon fruit.
Identification of the genes related to the TCA cycle
-
A total of 18 genes were identified to be involved in the TCA cycle during melon fruit development, including three PK, three MDH, three PCKA, two ACLY, two CS, two IDH, one PDHA, and one ACO (Fig. 6b). Among them, citric acid synthetase is positively correlated with citric acid content, and its expression is mainly higher in the middle and late stages of melon development. ACO, IDH, MDH, and other genes exhibited similar tendencies, and these genes were assigned to subcluster 3. The main function of ACO is to convert citric acid into isocitric acid, which is further converted to 2-ketoglutaric acid under the influence of IDH and participate in the synthesis of glutamic acid. Furthermore, the expression of four PK genes was higher in the early stages of melon development (M5D and M15D), which may offer pyruvate buildup for fruit growth. Simultaneously, the most common organic acids detected in metabolomics include citric acid, succinic acid, 2-oxoglutaric acid, levulinic acid, fumaric acid, trans-aconitic acid, isocitric acid, and D-alpha-hydroxyglutaric acid, among others (Fig. 6a). These organic acid levels are directly connected to transcriptional gene expression.
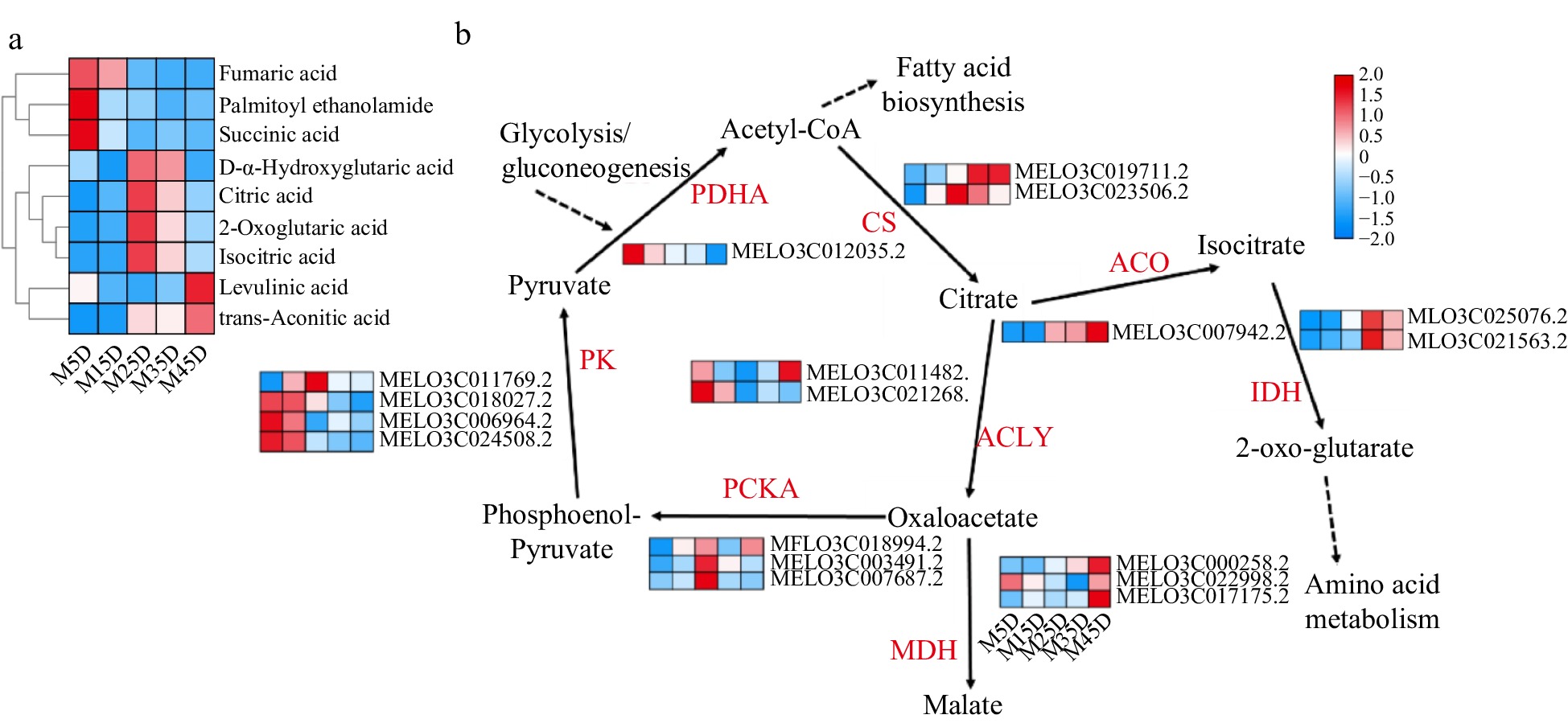
Figure 6.
Expression pattern of organic acid metabolic genes and identification in melon at five developmental stages. The development progression of gene expression from M5D to M45D is indicated in five box strings. PK, Pyruvate kinase; PDHA, pyruvate dehydrogenase; CS, Citrate synthase; ACO, aconitate hydratase; IDH, isocitrate dehydrogenase; ACLY, ATP citrate (pro-S)-lyase; PCKA, Phosphoenolpyruvate carboxykinase; MDH, Malate dehydrogenase.
Identification of genes involved in amino acid metabolism
-
Amino acids play an important role in assessing melon fruit quality and contribute to the development of distinct melon flavors[24]. Twenty-six genes related to amino acid metabolism were found in melon fruit development (Fig. 7a). AOC, as an enzyme that converts amines to amino acids, is actively expressed in the middle and late stages of development. The expression of most genes of cysteine synthase in M45D was significantly higher than that in other stages. The expression of genes synthesizing L-aspartate and β-alanine was also consistent with those mentioned above. In addition, some genes are active in early development, such as CYSE, THRC, SHMT, and TRPB. Analysis of metabolites corresponding to different clusters of gene-level clustering revealed that multiple amino acids were detected at different stages of melon development. It is mainly L-Tyrosine, L-Tryptophan, L-Phenylalanine, L-Isoleucine, L-Histidine, L-Cysteine, L-Aspartic Acid, L-(+)-Citrulline, L-(+)-Arginine, L-(−)-Methionine, D-(+)-Tryptophan, D-(−)-Glutamine, alpha-Aspartylphenylalanine, S-Lactoyl glutathione oxidized, Hexanoylglycine, Glycyl-L-Leucine, Gabapentin, etc. (Fig. 7b), Some of these amino acids are associated with the metabolic pathways of the identified genes, and their relative quantities correspond to specific developmental stages.
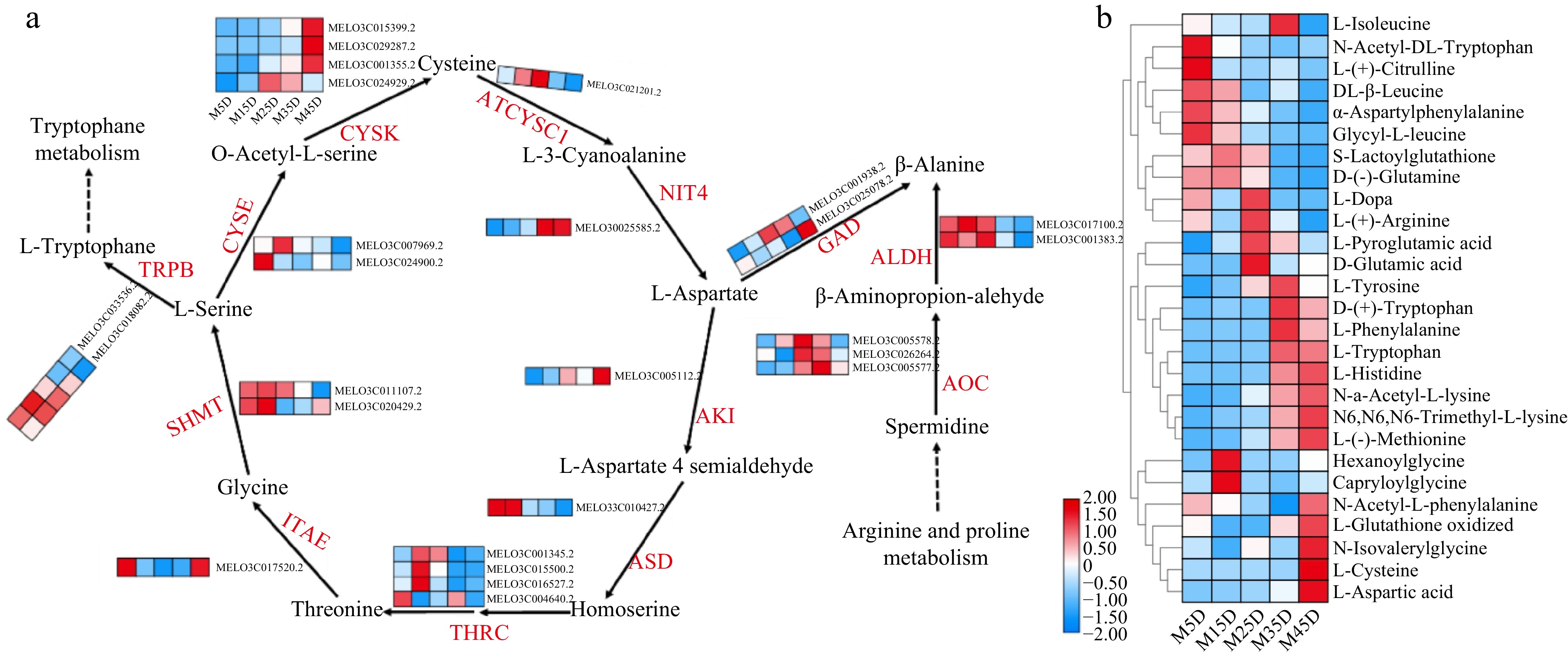
Figure 7.
Expression pattern of amino acid metabolic genes and identification in melon at five developmental stages. The development progression of gene expression from M5D to M45D is indicated in five box strings. ITAE, L-allo-threonine aldolase; THRC, threonine synthase; TRPB, tryptophan synthase; SHMT, serine hydroxymethyltransferase; CYSE, serine acetyltransferase; CYSK, cysteine synthase; ATCYSC1, L-3-cyanoalanine synthase; NIT4, bifunctional nitrilase/nitrile hydratase; AOC, Amine oxidase; ALDH, aldehyde dehydrogenase (NAD+); GAD, glutamate decarboxylase; AKI, aspartokinase; ASD, aspartate-semialdehyde dehydrogenase.
-
Fruit growth and ripening are closely associated with the accumulation of metabolites, leading to changes in fruit appearance, nutritional content, and flavor. As a result, it is crucial to identify the essential metabolic pathways and networks that contribute to enhancing fruit flavor. So far, although a large number of compounds have been identified as related to fruit quality, the metabolic pathways and regulatory mechanisms of these compounds remain largely unclear. In this study, through the comprehensive analysis of the metabolic and transcriptomic data of melon at five developmental and mature stages, 666 metabolites and 9377 corresponding genes were identified and distributed in five different clusters. By analyzing these data modules, the dynamic patterns of changes in melon metabolism and gene modules were revealed, and a metabolic regulatory network was constructed. The present findings not only confirmed the roles of known metabolic control genes and pathways using the provided gene and metabolite information, but more intricate metabolic networks governing fruit quality were also investigated. This was achieved through a combination of prior research knowledge and extensive analysis.
At present, transcriptomics, metabolomics, and other molecular methods have become important ways to study fruit quality metabolism[25, 26]. Similarly, this strategy was previously used to reveal key pathways in melon fruit flavor formation[24]. In this study, we further studied the quality metabolism of melon fruit was further studied and the metabolic pathways of soluble sugars, organic acids, and amino acids were established.
Sugar plays an important role in providing energy during the growth and development of melon fruit and contributes to their pleasant taste. As melon fruits continue to mature, the sugar content gradually increases[27]. The main soluble sugars found in melon fruits are glucose, fructose, and sucrose. It has been reported that the sugar content, composition, and ratio in melon fruits are the key factors determining fruit quality[28, 29]. Sucrose accumulation, which is a complex process in sugar metabolism, is an important factor in the increase of sugar content in fruits[30]. Research has shown that the enzymes that play an important role in sucrose metabolism mainly include sucrose synthase, sucrose phosphate synthase, acid invertase, and neutral invertase[31, 32]. In this study, 15 genes related to sugar metabolism and 10 major metabolites were identified through omics analysis. Among them, as an important enzyme for the distribution of photosynthetic products, sucrose phosphate synthetase is related to the synthesis of both sucrose and starch[33]. We found that the activity of this enzyme was higher in the early and late stages of melon development. This suggests that starch accumulation is inhibited and sucrose synthesis is promoted during the early and late stages of melon development. Correspondingly, the content of raffinose, which is a photosynthesizing product, was the highest at the end of melon development. Interestingly, we did not find sucrose in the metabolite identification. Only some sugars associated with sucrose accumulation were found, such as raffinose, mannose, and glucose. These phenomena may be related to sucrose synthesis in the subsequent melon post-ripening process, which needs further investigation. Three PYGs were identified, which are highly expressed during the middle stage of melon development and are associated with starch metabolism. The accumulation of mannose in the early stage of melon development and its subsequent decrease may be due to the action of 6-phosphate mannose isomerase. 6-phosphomannose isomerase catalyzes the formation of fructose-6P from D-mannose-6-phosphate using mannan as a substrate and participates in sucrose accumulation[34].
Organic acids are one of the indispensable factors determining the flavor and quality of fruits, and muskmelons are no exception. The main organic acids found in muskmelons include citric acid, and malic acid[35, 36]. These organic acids are primarily produced through the TCA cycle. In the TCA cycle, citric acid can catalyze the synthesis of oxaloacetate and acetyl CoA through CS; citric acid will be decomposed into isocitric acid under the action of aconitic acid synthase. Isocitric acid and ketoglutaric acid can be transformed by isocitric acid dehydrogenase (Fig. 6). Therefore, aconitic acid synthase and isocitric acid dehydrogenase can be regarded as enzymes encoding the citric acid pathway[37, 38]. In this study, the expressions of aconitate hydratase, isocitrate dehydrogenase, and citrate synthase were all higher during development, especially during late development, indicating that these enzymes were the key regulators of citric acid metabolism. Meanwhile, we identified citric acid but not malic acid in melon fruits at different stages of development. Only 2-isopropylmalic acid, a derivative of malic acid, was identified, which decreased first and then increased during the development of melon. This may be related to the formation of melon flavor substances. Citric acid increased first and then decreased during the growth period. It was correlated with CS. Pyruvate serves as a central node connecting sugar, acid, and amino acid metabolism. Phosphoenol-pyruvate is produced by the conversion of oxaloacetate through PCKA and is then converted to pyruvate by PK. Three PCKA and four PK genes were identified, which showed significant differences in different developmental stages. In addition, we identified three MDH genes. MDH are enzymes linked to malic acid biosynthesis in the fruit[39]. Although we did not identify malic acid in the metabolite identification, MDH genes related to malic acid metabolism were highly active in the late growth period of melon.
The amino acid route is one of the synthesis mechanisms for fruit fragrance, and the important enzymes for aroma component production are amino transferase and pyruvate dehydrogenase. These two enzymes transaminate amino acids to generate alpha ketoacids, which are then decarboxylated or dehydrogenated to form aldehydes. These enzymes catalyze the transamination of amino acids to generate alpha-ketoacids, which are subsequently decarboxylated or dehydrogenated to form aldehydes. Under the action of dehydrogenase, aldehydes can further react to form alcohols and esters, which contribute to the overall aroma profile of melon fruits[40, 41]. Therefore, amino acid content was significantly correlated with melon quality. In this study, the pathway of amino acid metabolism in melon fruit was preliminarily established, and 26 genes related to amino acid metabolism were excavated. Several amino acids were identified by metabolomics, among which tyrosine, tryptophan, leucine, and phenylalanine were related to melon aroma synthesis. phenylalanine acts as a precursor for the production of volatile phenolic substances. In this pathway, branched aliphatic alcohols, aldehydes, and esters are mainly generated[42]. In this study, the pathway of amino acid metabolism in melon fruit was preliminarily established, and 26 genes related to amino acid metabolism were excavated. Several amino acids were identified by metabolomics, among which tyrosine, tryptophan, leucine, and phenylalanine were related to melon aroma synthesis. In the present study, these amino acids showed significant differences in the later stages of melon fruit development, suggesting that amino acids provide sufficient precursor substances for melon flavor formation during post-ripening. Studies have shown that glutamate and aspartic acid are derived from alpha-ketoglutaric acid and oxaloacetic acid in the TCA cycle. These amino acids serve as precursors for the biosynthesis of other amino acids through various biochemical reactions[43]. It was found that glutamate increased first and then decreased during melon development, reaching its peak at M25D. Aspartic acid increased gradually with the late growth and development of melon. In addition, three amino oxidase genes were identified that could convert amine compounds into aldehydes and then convert them into amino acids under aldehyde dehydrogenase (Fig. 7). This may be another pathway of amino acid synthesis that needs to be further explored. In summary, a number of genes related to amino acid metabolism were identified, the dynamic changes of various amino acids during the growth and development of melon were also revealed. However, the metabolic mechanism of some amino acids is still unclear, and further analysis may be needed in combination with biochemical indicators such as proteomics or amino acid content.
-
The transcriptome and metabolomics of five melon fruit growth and development phases were investigated. Through comprehensive analysis, the metabolic pathways of key metabolites were identified, which provided a basis for the subsequent improvement of fruit quality and further study of the functions of these key metabolites. This study provides insight into the major metabolic changes in melon fruit development. The key enzymes in glucose metabolism, organic acid metabolism, and amino acid metabolism were identified. The major metabolic markers and their synthetic metabolic pathways were analyzed. Furthermore, the amino acid pathway's primary role in melon aroma production was explored. By elucidating the major metabolic changes and pathways in melon fruit development, our study offers valuable insights for managing fruit quality and provides a framework for investigating important metabolite metabolic pathways. This knowledge can be leveraged to enhance fruit quality and further our understanding of the underlying mechanisms governing melon fruit metabolism.
-
The authors confirm contribution to the paper as follows: study conception and design: Wang C, Wang F; data collection: Liu F, Shen Q, Jia B; analysis and interpretation of results: Shao X, He W, Fan Y; draft manuscript preparation: Shao X, Liu F. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and analyzed during the current study are available from the corresponding author on reasonable request.
This study was funded by China Postdoctor (No. 299580), Modern-Agroindustry Technology Research System (CARS-25), the earmarked fund for XinJiang Agriculture Research System (XJARS-06), Science & Technology Department of Xinjiang Uygur Autonomous Region (2022A02006-1) and the special project for basic scientific activities of non-profit institutes supported the government of Xinjiang Uyghur Autonomous Region (KY2021118 and KY2020108).
-
The authors declare that they have no conflict of interest.
-
Authors contributed equally: Xupeng Shao, Fengjuan Liu
- Supplemental Table S1 Metabolite expression was detected in the metabolome.
- Supplemental Table S2 Annotation of metabolite KEGG function.
- Supplemental Table S3 Transcriptome raw data quality comparison.
- Supplemental Fig. S1 Heat map of different genes at 5 developmental stages in melon.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of China Agricultural University, Zhejiang University and Shenyang Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Shao X, Liu F, Shen Q, He W, Jia B, et al. 2024. Transcriptomics and metabolomics reveal major quality regulations during melon fruit development and ripening. Food Innovation and Advances 3(2): 144−154 doi: 10.48130/fia-0024-0013 

Transcriptomics and metabolomics reveal major quality regulations during melon fruit development and ripening
- Received: 03 April 2024
- Revised: 10 May 2024
- Accepted: 13 May 2024
- Published online: 30 May 2024
Abstract: Studying the metabolic patterns underlying the key quality traits during the growth and development of melon is very important for the quality improvement and breeding of melon fruit. In this study, we employed transcriptomics and metabolomics to analyze the primary metabolic changes occurring in melon ('Xizhoumi 25') across five growth and development stages. We identified a total of 666 metabolites and their co-expressed genes, which were categorized into five different metabolic and gene modules. Through the analysis of these modules, the main metabolic pathways during the growth and development of melon were demonstrated from a global perspective. We also discussed the contribution of sucrose accumulation, the TCA cycle, and amino acid metabolism to the quality and flavor of melon. Enzymes related to amino acid metabolism were proposed, including Amine oxidase (AOC), aldehyde dehydrogenase (ALDH), tryptophan synthase (TRPB), etc. These results and data can provide new insights for further study on the metabolic regulation of melon quality and improve fruit quality.
-
Key words:
- Melon /
- Transcriptomics /
- Metabolomics /
- Development /
- Metabolic network