










-
Sugars are predominant carbon and energy sources and support plant vegetative and reproductive growth[1]. Transport of sugars across the plant bio-membrane needs the assistance of specific transporters[1,2]. These transporters act as bridges that mediate the distribution of sugars between source–sink organs, which is critical for sugar homeostasis and the cellular exchange of sugar efflux in multicellular organisms[1,3,4].
SWEETs (Sugars Will Eventually be Exported Transporter) and SUTs (sucrose transporters), MSTs (monosaccharide transporters) are the main known sugar transporters in eukaryotes[5] . Unlike SUTs and MSTs, the relatively newly reported sugar transporter SWEETs are pH-independent transporters. SWEETs play important roles in phloem transport and act as bidirectional transmembrane transporters of sugars along the concentration gradient[3,4]. AtSWEET1 was first identified as a glucose transporter with clear functional characterization[2]. In addition, the SWEET multi-gene family was identified and classified into four clades and the functional divergence of these paralogs were also revealed in Arabidopsis at the same time[2]. For example, Clade II AtSWEET8 contributes to pollen viability and Clade III AtSWEET15 is involved in leaf senescence. Thereafter, another SWEET in Arabidopsis Clade III AtSWEET9 was characterized as an important transporter involved in nectar secretion[6]. Besides the SWEET family in Arabidopsis, the SWEET gene family has been identified in many plants including Tea (Camellia sinensis, Cs), tomato (Solanum lycopersicum, Sl), wheat (Triticum aestivum, Ta) barrel medic (Medicago truncatula, Mt), cabbage (Brassica rapa, Br), daylily (Hemerocallis fulva, Hf), grapevine (Vitis vinifera, Vv), rice (Oryza sativa, Os) and poplar (Populus trichocarpa, Pt and P. alba × P. glandulosa, Pag)[7−13]. These SWEET homologs belong to the MtN3/saliva family and consist of seven α-helical transmembrane domains (TMs): a tandem repeat of three transmembrane domains (TMs) connected with a linker-inversion TM[2,14].
SWEETs participate in various biological processes including development, flowering, stress responses and plant-pathogen interaction in plants[4,15]. In addition, different SWEET gene family members show functional divergence or redundancy. In Arabidopsis, AtSWEET8 and 13 support pollen development; AtSWEET11 and 12 provide sucrose to the SUTs for phloem loading and play distinct roles in seed filling; and AtSWEET9 is essential for nectar secretion[2−4,6,16]. BrSWEET9 in Brassica rapa was also reported to be involved in nectar secretion[17]. Overexpression of PagSWEET7 promotes secondary growth and xylem sugar content[12]. OsSWEET11 and 15 have functions affecting pollen development and are key players in seed filling in rice[18]. SWEET homologs also play important roles in abiotic stress responses. Overexpression of AtSWEET16 promotes freezing tolerance in Arabidopsis[19] while AtSWEET11 and 12 mutants exhibit greater freezing tolerance[20]. AtSWEET15 could be induced by various abiotic stresses including osmotic, drought, salinity, and cold stresses and overexpression of AtSWEET15 results in transgenic plants with hypersensitiveness to cold and salinity stresses[21]. CsSWEET1a and CsSWEET16 were also reported to mediate freezing tolerance[22,23]. Recently, more studies have shown that SWEETs are involved in plant-pathogen interaction and are known as susceptibility (S) genes, acting as targets of effector proteins during host–microbe interactions in many plant species[15]. GhSWEET10, is induced by Avrb6, a transcription activator-like (TAL) effectors from Xanthomonas citri subsp. Malvacearum (Xcm) and is responsible for maintaining virulence of Xcm avrb6 and the cotton susceptibility to infections[24]. OsSWEET11–15 belonging to clade III in rice have been shown to be induced by TAL effector from Xanthomonas oryzae and support pathogen growth[25]. In contrast, some SWEETs could also function as resistance genes. Overexpression of IbSWEET10 can promote resistance to F. oxysporum in sweet potato[26]. Mutation of AtSWEET2 resulted in increased susceptibility to the root necrotrophic pathogen Pythium irregulare[27].
Mulberry (Morus spp., Moraceae) is a traditional economic crop plant and a new beverage plant. In addition, its fruits are rich in nutrient and bioactive components and the ripening process of mulberry fruits along with sugar accumulation and distribution. Mulberry suffers various abiotic stresses and the disasterous fungal disease sclerotiniose which bursts at the early stage of mulberry fruit development[28−30]. Mulberry fruits with sclerotiniose lose their color and flavor and turn pale instead of ripening. C. shiraiana is the dominant causal agent of mulberry sclerotiniose in China, and it results in hypertrophy sorosis sclerotiniose. SWEETs as the important transporters involved in sugar homeostasis are expected to be involved in mulberry fruit development and interaction with sclerotiniose pathogens. However, to date, few studies on SWEETs have been reported in mulberry, although the SWEET gene family may play important roles in mulberry fruit development and responses to abiotic and biotic stresses. Mulberry genome information has been released successively since the Morus notabilis genome was reported in 2013[31]. The chromosome-level genome of M.alba (Ma) was released by Jiao et al. and the genome of M. yunnanensis was recently released by Xia et al.[32,33]. Released genome information makes it possible to perform genome-wide characterization of the SWEET gene family in mulberry. In the present study, a total of 24 SWEET genes were identified in the Morus alba genome and their phylogenetic classification, conserved motifs, gene structures, distribution on chromosomes, cis-elements in promoter regions and tissue expression profile were revealed. In addition, the responses of MaSWEETs to various abiotic stresses and sclerotiniose pathogen infection were also detected. MaSWEET1a was functionally characterized as a negative regulator which increased the mulberry susceptibility to C. carunculoides infection.
-
The xylem, phloem, fruits at four different developmental stages (S0, inflorescence; S1, green fruits; S2, reddish fruits; S3, purple fruits)) and diseased fruit infected with C. shiraiana of Morus atropurpurea variety Zhongshen 1 (Mazs) were collected from the National Mulberry Genebank (NMGB) in Zhenjiang, China for expression profiling. Seedlings of the M. alba var. Fengchi and tobacco (Nicotiana Benthamiana) were grown in a chamber at 22 °C with a 16/8 day/night cycle and 40%–60% humidity. C. shiraiana was provided by Professor Zhao and was cultured in potato dextrose agar (PDA) medium.
Tobacco at the four-leaf stage was used for transient overexpression. M. alba var. Fengchi seedlings at the four-euphylla stage were used for virus-induced gene silencing (VIGS). Four-week-old seedlings with similar growth conditions (~12–15 cm high) were used for treatments under different abiotic stresses. Detailed information for abiotic stress treatments were reported in our previous study[34]. All the above samples were immediately frozen in liquid nitrogen after collection and then stored at −80 °C until use. Three biological replicates were performed for each experiment.
Identification of the SWEET gene family in Morus alba
-
The M. alba genome sequences (.fasta) and annotation file (.gff) were generously provided by Professor Jiao, who released this genome information. The Hidden Markov Model (HMM) profiles of the SWEET domain (PF03083) were downloaded from the Pfam database (
http://pfam.xfam.org/ ) and used to search the candidate SWEET proteins in the M. alba proteome with HMMER software. In addition, the protein sequences of AtSWEETs, OsSWEETs and PtSWEETs were downloaded from TAIR (www.arabidopsis.org/ ), TIGR (http://rice.plantbiology.msu.edu/ ) and phyto-zome (https://phytozome-next.jgi.doe.gov/ ) respectively, and used as queries to search against the M. alba proteome. The Toolbox for Biologists v1.098774[35] was used to analyze the sequence length, molecular weight and theoretical isoelectric point (pI) values of each MaSWEET protein. The distributions of TM helices were predicted by the TMHMM Server v. 2.0 (www.cbs.dtu.dk/services/TMHMM ). Prediction of subcellular localization of MaSWEET proteins using the online Tool WoLF PSORT (www.genscript.com/wolf-psort.html )[36].Chromosomal location and synteny analysis of MaSWEETs
-
Chromosome location information of MaSWEETs was extracted based on the Morus alba genome annotation file. Tbtools v1.098774 were used to identify syntenic blocks and tandem duplication events using default parameters[37,38]. The results were visualized using Tbtools v1.098774 and both the tandem duplication and block duplication gene pairs were marked.
Sequence alignment and motif analysis
-
MaSWEETs were aligned using clustal W assembled in MEGA11.0. The alignment result was exported and manually speculated for scanning the MtN3 repeats. The online MEME Suite version 5.5.0 was used to identify 7 conserved motifs from 24 amino acid sequences of SWEET genes in Morus alba. The Hidden Markov Model (HMM) profiles of the SWEET domain (PF03083) were downloaded from the Pfam database.
Gene structure and promoter analysis of MaSWEETs
-
The gene structure of each MaSWEET was displayed based on the genome sequence and its annotation file using Gene Structure View assembled in Tbtools v1.098774. The upstream 2000 bp sequences were extracted for in silico promoter region analysis. Cis-acting elements were predicted using PlantCARE (
http://bioinformatics.psb.ugent.be/webtools/plantcare/html/ ).Phylogenetic analysis of MaSWEETs
-
A neighbor-joining (NJ) phylogenetic tree was constructed using full-length SWEETs protein sequences from A. thaliana, P. trichocarpa, O. sativa and M. alba using MEGA11.0[26] with JTT + G model and bootstrap test with 1000 replicates.
RNA extraction and RT-qPCR analysis
-
RNA extraction and cDNA synthesis were performed as in our previous report using Plant RN52 Kit (Aidlab, Beijing, China) and PC54-TRUEscript RT kit (Aidlab, Beijing, China) according to the manual[39]. RT-qPCR (quantitative real-time PCR) was performed to validate the expression patterns of MaSWEETs in different tissues, fruit development stages and stresses using ABI StepOnePlus™ Real-Time PCR System (USA). The primers are available in Supplemental Table S1. Actin was used as a reference gene[40]. Graphpad Prism8.0 was used to visualize the RT-qPCR results and to perform T-test and ANOVA. P value < 0.05 was marked as significant. At least three individuals were used and three technical replicates respectively were performed for RT-qPCR.
Transient overexpression of MaSWEET1a in Nicotiana Benthamiana
-
The recombinant plasmids pNC-1304-35S:MaSWEET1a were constructed using nimble cloning[41]. Both recombinant plasmids pNC-1304-35S: SWEET1a and empty vector pNC-Cam1304-35SMCS, as the negative control, were transformed into Agrobacterium tumefaciens GV3101 and then transferred into N. benthamiana leaves via Agrobacterium-mediated transient transformation, as previously reported[41]. Overexpression of MaSWEET1a was determined using RT-qPCR by comparing the expression levels of target genes in transgenic plants with those in the negative controls.
Obtaining MaSWEET1a/b VIGS Transgenic Mulberry
-
Virus-induced gene silencing (VIGS) was used to obtain MaSWEET1a/b down-regulated mulberry, in accordance with our previous report[42]. Agrobacterium tumefaciens containing recombinant plasmids pTRV2-MaSWEET1a/b, pTRV1 and pTRV2 (negative control) were cultured in transient transformation buffer and then transferred into mulberry leaves by means of pressure injection. Ten independent mulberry plants were injected. The knock-down efficiency for the target genes was determined by RT-qPCR 15 d after injection by comparing the transgenic plants with the negative controls and wild types.
Estimation of plant resistance to C. shiraiana infection
-
Cell death symptoms and the growth condition of C. shiraiana were recorded to estimate the resistance of transgenic plants to C. shiraiana infection[43,44]. C. shiraiana was inoculated at 2 d after infiltration in tobacco and at 10 d after infiltration in mulberry. The cell death symptoms were photographed after inoculation until the sclerotia appeared. The results are representative of at least three biological replicates.
-
A total of 24 SWEET homologs were identified based on the genome information of M. alba and named according to their orthologs in A. thaliana, P. trichocarpa or V. vinifera[45]. These MaSWEETs encode proteins ranging from 197 aa to 304 aa with molecular weight from 21.45 to 34.12 kDa and theoretical isoelectric points from 7.16 to 9.61 (Table 1). Subcellular localization prediction of these MaSWEETs showed that most of them (18/24) distributed on membrane structures such as plasma membrane (PM), tonoplast membrane (TM) and chloroplast thylakoid membrane (CTM). Phylogenetic analysis of MaSWEETs and SWEETs from model plants such as A. thaliana, P. trichocarpa and O. sativa showed that four clades were formed by these SWEETs (Fig. 1). According to previous studies, SWEETs in plants were generally classed as four phylogenetic clades which is in agreement with our results[8]. Major MaSWEETs (10/24) together with AtSWEET1, 2 and 3 belong to clade I. Clade II and IV contain five MaSWEETs each and Clade III contains four MaSWEETs (Fig. 1 and Table 1).
Table 1. SWEET gene family in Morus alba.
Clade Gene name Accession no. Gene ID CDS Size
(bp)Protein physicochemical characteristics TMHs Subcellular MtN3/Saliva (PQ-Loop Repeat) Length (aa) MW (kDa) pI Aliphatic index Localization* Domain Position I MaSWEET1a XM_024170697.1-0 M.alba_G0012049 729 242 26.55 9.61 113.51 7 CTM 6-94, 131-209 I MaSWEET1b XM_024170698.1-0 M.alba_G0012049 678 225 21.45 9.51 115.74 6 CTM 1-49, 86-164 I MaSWEET2a XM_024163709.1-0 M.alba_G0019244 708 235 25.94 8.39 129.79 7 CTM 18-104, 137-221 I MaSWEET2b XM_024164777.1-0 M.alba_G0010863 705 234 25.92 8.98 122.86 7 TM 17-101, 137-218 I MaSWEET2c XM_024163707.1-0 M.alba_G0019244 774 257 28.58 8.58 132.33 7 PM 54-126, 159-243 I MaSWEET2d XM_024163712.1-0 M.alba_G0019244 681 226 25.27 8.22 128.89 6 EX 23-95, 128-212 I MaSWEET2e XM_024163708.1-0 M.alba_G0019244 729 242 26.72 8.49 128.06 7 CTM 39-111, 144-228 I MaSWEET2f XM_024163703.1-0 M.alba_G0019244 777 258 28.8 8.49 132.56 8 PM 41-127, 160-244 I MaSWEET2g XM_024163711.1-0 M.alba_G0019244 684 227 25.49 7.62 129.16 7 PM 10-96, 129-213 I MaSWEET3 XM_010099554.2-0 M.alba_G0003063 783 260 29.01 8.89 115.73 7 PM 9-98, 132-216 II MaSWEET4a XM_010091939.1-0 M.alba_G0009276 735 244 27.45 9.28 109.39 7 CTM 10-98, 134-218 II MaSWEET4b XM_010113461.2-0 M.alba_G0001536 738 245 27.4 8.98 122.08 7 EX 11-97, 134-216 II MaSWEET5 XM_024168739.1-0 M.alba_G0018295 711 236 26.64 7.63 120.93 7 CY 10-93, 131-127 II MaSWEET7a XM_010108966.2-0 M.alba_G0005110 774 257 28.32 9.57 128.56 7 CTM 11-95, 134-218 II MaSWEET7b XM_010108964.1-0 M.alba_G0005109 789 262 28.99 9 124.96 7 CTM 10-97, 134-218 III MaSWEET10 XM_010095631.2-0 M.alba_G0018016 888 295 33.12 8.86 120.31 7 CTM 11-96, 132-216 III MaSWEET11a XM_010114440.2-0 M.alba_G0016901 804 267 29.63 9.47 124.08 7 CTM 11-99, 135-220 III MaSWEET11b XM_010095633.2-0 M.alba_G0018015 915 304 34.12 7.57 112.89 7 CTM 12-99, 134-219 III MaSWEET15 XM_010092381.2-0 M.alba_G0006767 885 294 33.42 7.16 109.01 7 CTM 12-99, 133-219 IV MaSWEET16 XM_024167733.1-0 M.alba_G0014617 909 302 33.2 9.08 114.27 7 CTM 20-92, 129-212 IV MaSWEET17a XM_024171451.1-0 M.alba_G0014614 708 235 26.46 8.71 119.87 5 CY 5-78, 116-198 IV MaSWEET17b XM_024167902.1-0 M.alba_G0014613 753 250 28.05 8.71 120.88 6 PM 8-93, 131-213 IV MaSWEET17c XM_024171286.1-0 M.alba_G0008195 720 239 26.72 8.94 111.72 7 CY 6-92, 129-212 IV MaSWEET17d XM_024171287.1-1 M.alba_G0008196 723 240 27 9.43 122.67 7 CTM 6-92, 127-213 * The subcellular localizations were predicted by WoLFPSORT. PM, plasma membrane; EX, extracellular; CY, cytoplasmic; TM, tonoplast membrane; CTM, chloroplast thylakoid membrane. 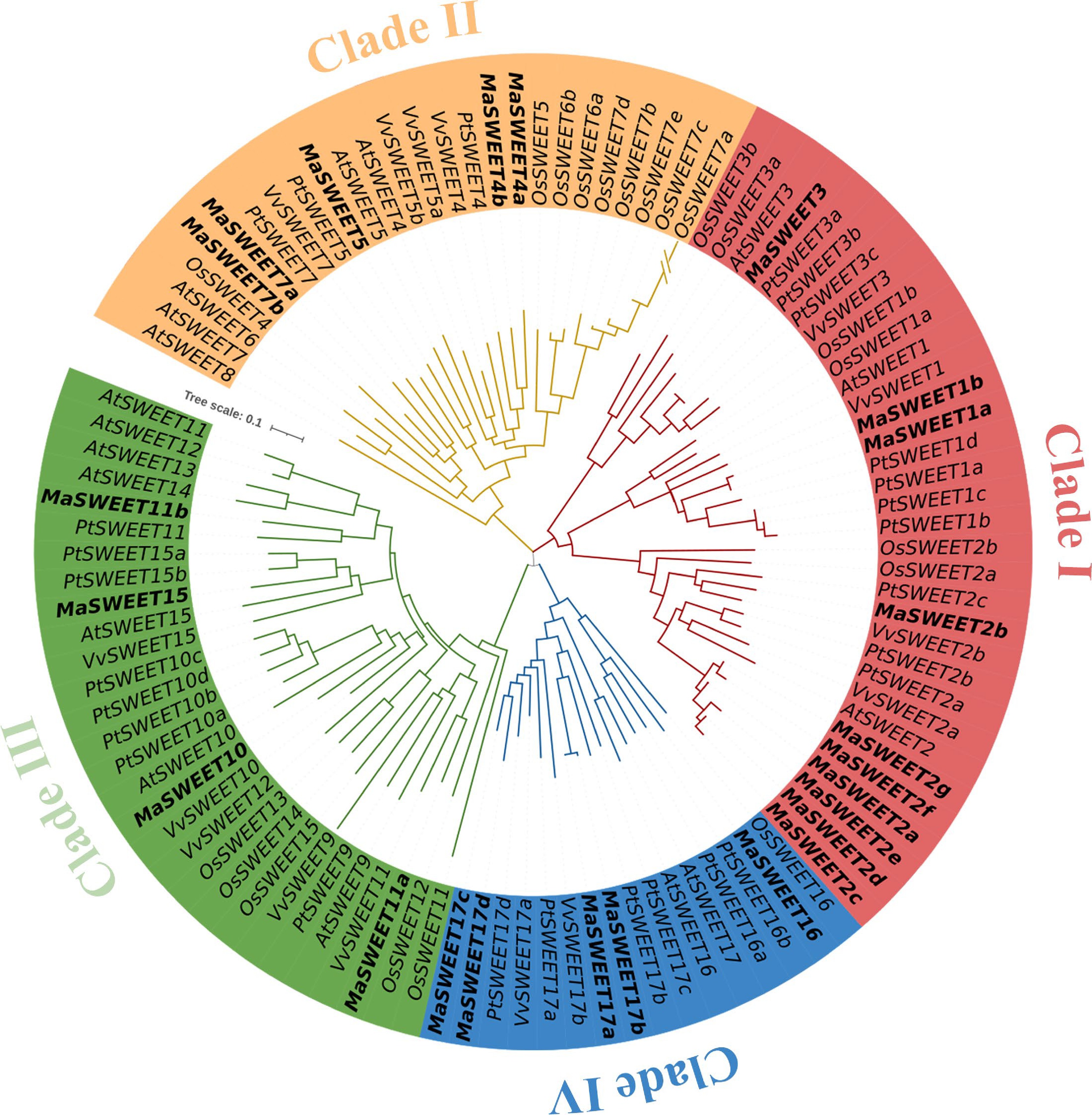
Figure 1.
Phylogenetic relationships of the SWEET family genes in Arabidopsis, Oryza sativa, Populus, Vitis vinifera, and Morus alba. The sequences of the 104 SWEET proteins from the above four plant species were aligned by Clustal Omega, and the phylogenetic tree was constructed by the MEGA 11.0 using the NJ method with 1000 bootstrap replicates. The proteins from Arabidopsis, Oryza sativa, Populus, Vitis vinifera, and Morus alba are indicated with the prefixes of At, Os, Pt, Vv, and Ma, respectively.
Chromosomal location and gene duplication
-
MaSWEETs distributed on 12 chromosomes except chromosome 1 and 3. Chromosome 5 occupied five MaSEETs which formed a gene cluster. Chromosome 5 is the chromosome that had the most SWEETs and the following is chromosome 6 and 8 which had four MaWSEETs each. There was only one MaSWEET locating on chromosome 4 (Fig. 2). In addition, there were three MaSWEETs on chromosome 2 and 7 respectively and two MaSWEETs on chromosome 1 and 5 respectively. Gene duplication including block duplication and tandem duplication is the main cause for gene family expansion. Tandem duplications were found on chromosome 2, 6 and 12 (linked by red lines in Fig. 2). It is also interesting to find several possible gene clusters such as MaSWEET1a-b on chromosome 5, MaSWEET2a-g on chromosome 9, MaSWEET7 a-b on chromosome 13, MaSWEET17a-b on chromosome 12 and MaSWEET17c-d on chromosome 6. Two gene pairs (MaSWEET4b/MaSWEET5 and MaSWEET11b/ MaSWEET15) resulting from block duplications were also marked (linked by black lines in Fig. 2).
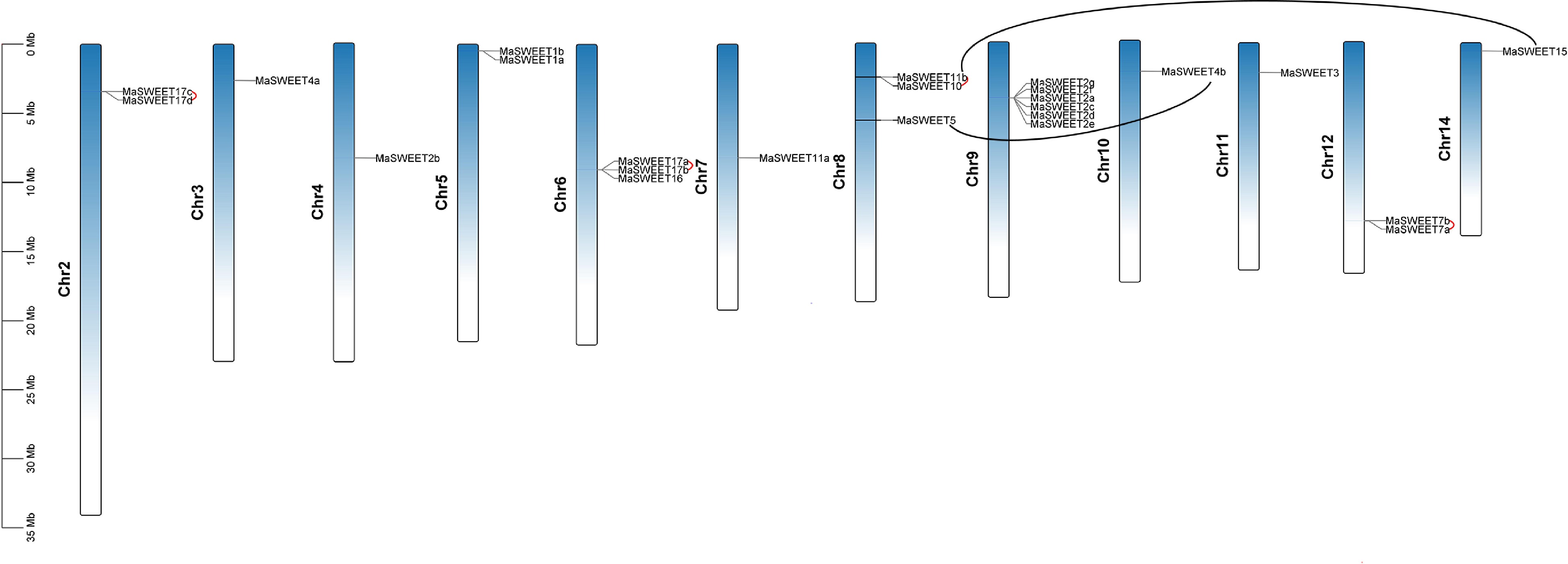
Figure 2.
Distribution of MaSWEET genes in Morus alba chromosomes. The tandem gene pairs are linked by red lines. The block duplications gene pairs are marked by black lines. The scale is provided in megabase (Mb).
Sequence analysis of MaSWEETs
-
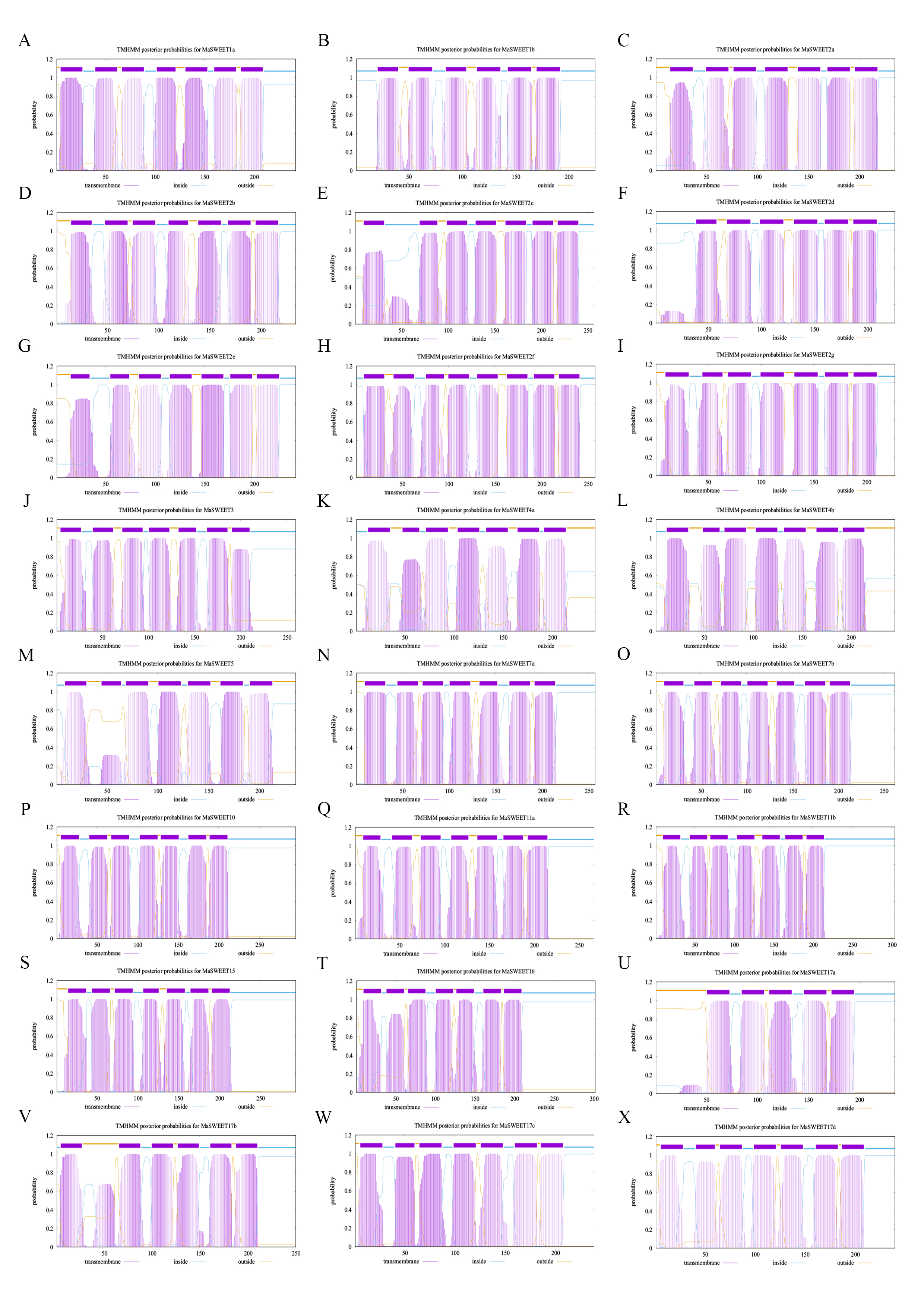
MaSWEETs always located on membrane structures with transmembrane domains. The prediction results of MaSWEETs using DeepTMHMM showed that most MaSWEETs posed seven types of transmembrane helices (TMH) named TMH1-7 (Table 1, Supplemental Fig. S1). Alignment and conserved motif analysis showed that almost all MaSWEETs kept the conserved TMH and active sites Tyr and Asp indicating by red full triangles (Fig. 3). The active residues Tyr and Asp were reported to be involved in forming hydrogen bonds to ensure sugar transport activity[14]. In addition, all MaSWEETs except MaSWEET4a had a conserved Ser in each triple helix bundle (THB) which can be phosphorylated and is important for SWEET activity (Fig. 3). MaSWEET4a replaced Ser with Thr at the first Ser phosphorylation site between TMH1 and TMH2 which may also retain similar activity as Thr is also a common phosphorylation site. All MaSWEETs retained the conserved second Ser phosphorylation site between TMH5 and TMH6 (Fig. 3).

Figure 3.
Multiple sequence alignment of MaSWEET proteins. The positions of the TMHs are underlined. The positions of the active sites of tyrosine (Y) and aspartic acid (D) are indicated by red triangles. The conserved serine (S) phosphorylation sites are indicated by blue triangles.
Gene organization and promoter analysis of MaSWEETs
-
MaSWEETs gene structures were identified based on the annotation information of the M. alba genome. In summary, there were six MaSWEETs with six introns, 12 MaSWEETs with five introns and five MaSWEETs with four introns (Fig. 4). In addition, genes clustered together based on phylogenetic analysis are likely to show similar gene structures and length. For example, MaSWEET2a, c, d, e, f and g with six introns, MaSWEET7a and MaSWEET7b with four introns, and MaSWEET17a and MaSWEET17b with four introns.
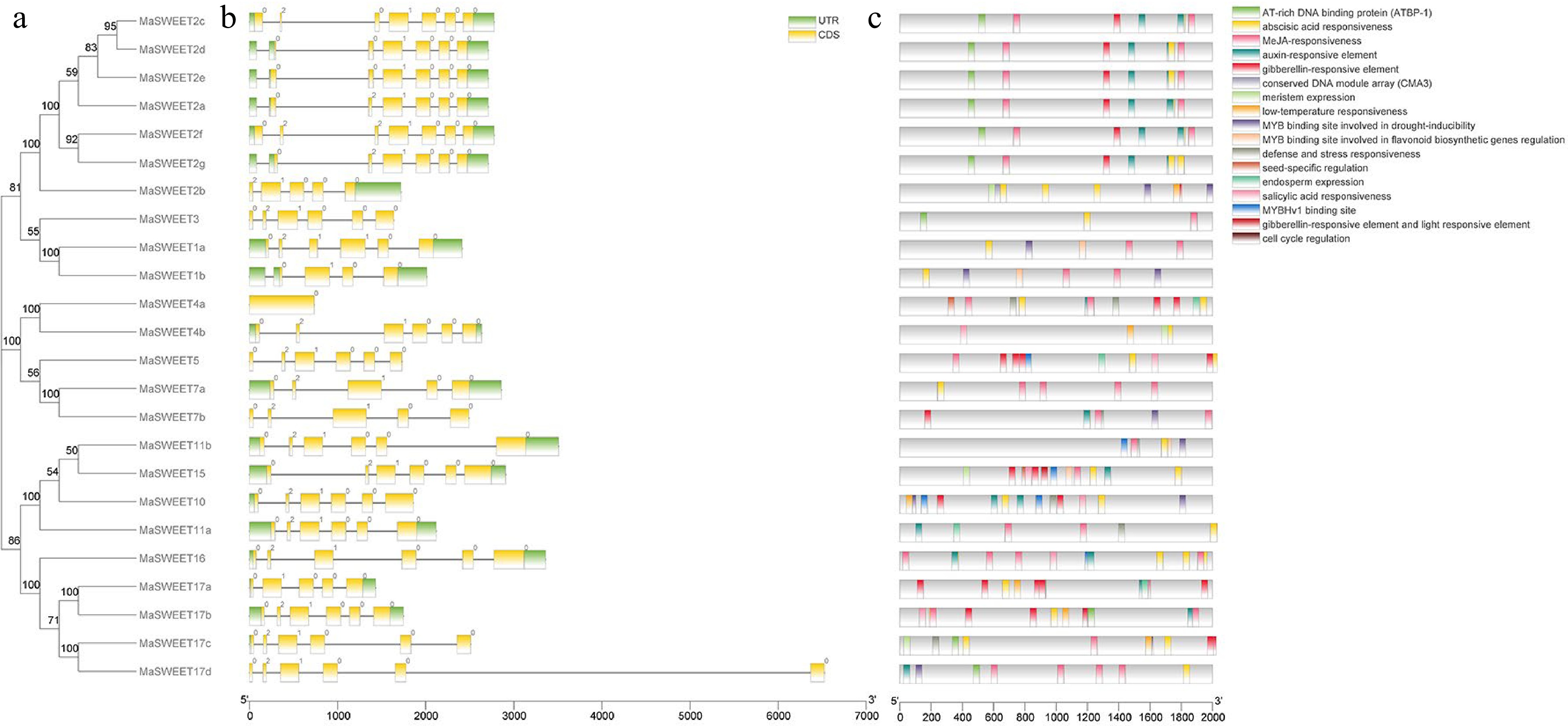
Figure 4.
Gene organization of MaSWEETs and cis-elements in promoter regions of MaSWEETs. (a) Phylogenetic tree using 24 MaSWEETs. (b) Exon/intron structures of Morus alba L. SWEETs. (c) Cis-element distribution in the promoter regions of MaSWEETs.
Promoter region analysis of MaSWEETs indicated the possible function of MaSWEETs in response to hormones and environment stimulus. Among all the 22 types of cis-elements identified, most of them are light response elements accounting for 44% of the total elements (Supplemental Table S2). In addition, hormone response elements were also widely identified in the promoters of MaSWEETs (Fig. 4c). Most MaSWEETs had abscisic acid (ABA), salicylic acid (SA) or methyl jasmonate (MeJA) related response elements in their promoter regions. Especially, MaSWEET1a-b, MaSWEET16, MaSWEET17a-d had cis-elements involved in response to five types of hormones (ABA, SA, MeJA, auxin and gibberellins). Several Myb binding cis-elements were also identified in promoter regions of MaSWEET2a and MaSWEET10 (Fig. 4c, Supplemental Table S2).
Expression profile of MaSWEETs in different tissues of mulberry
-
The tissue or organ expression profiles of MaSWEETs were revealed. The MaSWEETs with high sequence identity (> 91%) are hard to distinguished by RT-qPCR and were determined by common primers to reveal their total transcription levels. MaSWEETs in phylogenetic clade I showed quite similar expression patterns with highest expression levels in leaf and relatively higher expression levels in early stages (S0 and S1) of fruit development except MaSWEET1a/b (Fig. 5a−d). MaSWEET1a/b showed higher expression levels in fruits during whole fruit development with highest expression level in fruit at S0 stage (Fig. 5a). However, MaSWEETs (MaSWEET10, 11a-b, and 15) in phylogenetic clade III showed preferential expression in fruits especially at the late stages (S2 and S3) (Fig. 5j−m). MaSWEETs in phylogenetic clade II had different expression patterns in different tissues or organs. MaSWEET4a and b showed highest expression level in fruit at the S1 stage while MaSWEET5 showed obviously preferential expression in xylem (Fig. 5e−g). MaSWEET7a and b showed similar expression pattern with MaSWEET2 cluster and MaSWEET3 from phylogenetic clade I. MaSWEETs in phylogenetic clade IV showed similar expression pattern with highest expression levels in leaf (Fig. 5b, d, h, i). MaSWEET16 also had higher expression in fruit with similar expression level at four different development stages (Fig. 5n).
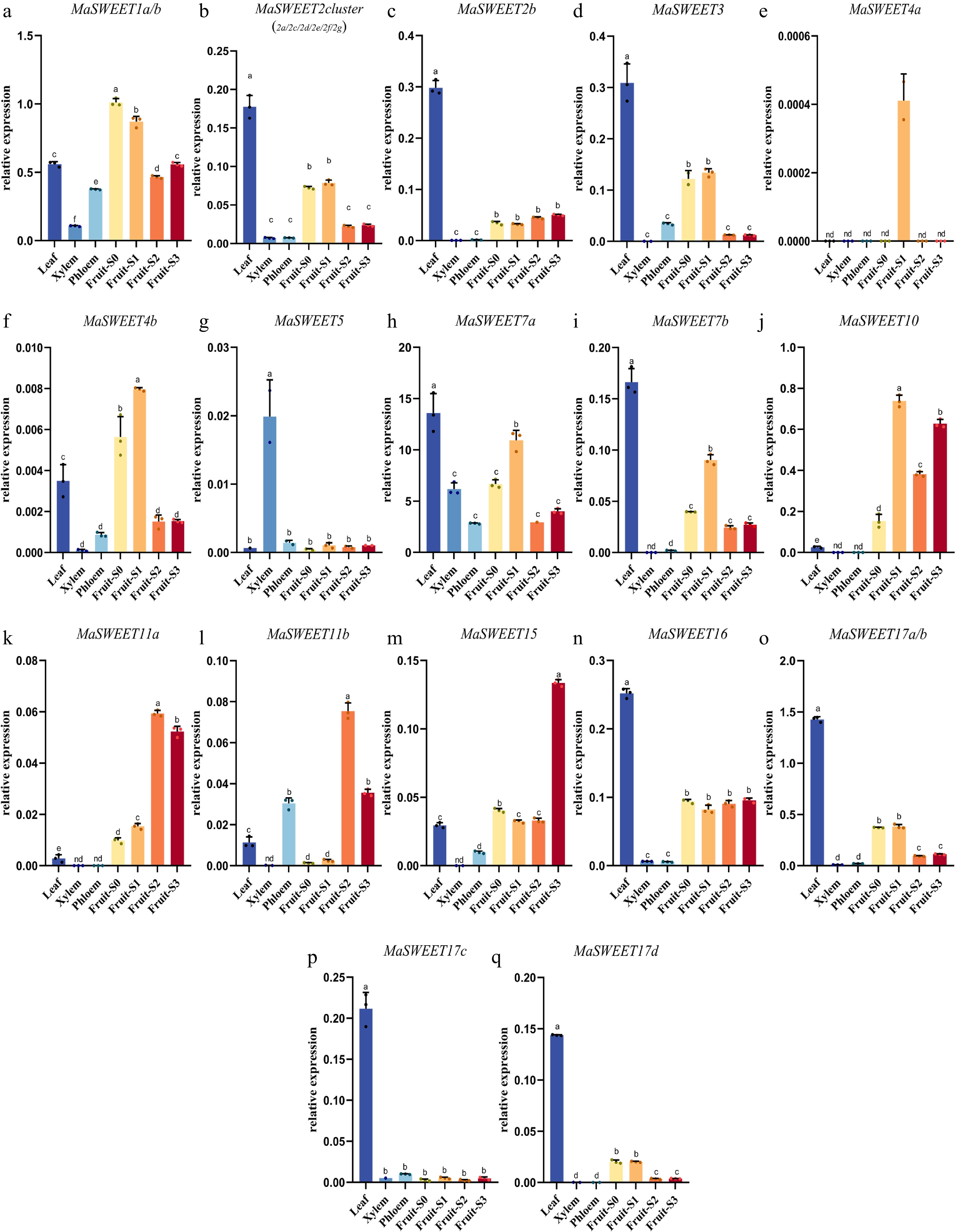
Figure 5.
Transcript levels of MaSWEETs in leaves, xylem, phloem, and different development stages of fruit. Three technical replicates were analyzed. Error bars represent SE. Different letters indicate statistically significant differences (Duncan's test, p < 0.05).
Transcription-level responses of MaSWEETs to various stresses
-
Mulberry sclerotiniose is a fungal disease resulting from fungal pathogen infection. Most (20/24) MaSWEETs showed positive or negative responses to the fungal infection. MaSWEET1a/b, MaSWEET2 cluster, MaSWEET4b, and MaSWEET17 a-d showed a significant decrease of expression levels in diseased fruits with sclerotiniose compared with the expression levels in healthy fruits (Fig. 6). In contrast, MaSWEET2b, MaSWEET3, MaSWEET7b, MaSWEET10 and MaSWEET11a-b showed significant increases of expression levels in diseased fruits. MaSWEETs also played roles in response to various abiotic stresses including drought, water logging, cold and high temperature. MaSWEET1a/b showed a positive response to drought with significant increasing expression levels while other clade I MaSWEETs, MaSWEET2b and 3 significantly decreased their expression level under detected abiotic stresses (Fig. 7a−d). In contrast, MaSWEET16 significantly increased its expression level under detected abiotic stresses. MaSWEET4a-b, MaSWEET5 in phylogenetic clade II and MaSWEET11a-b in clade III showed similar response patterns with a significant increase of expression levels in response to low temperature (4 °C), high temperature (40 °C) or drought (Fig. 7e−g). MaSWEET15 showed high sensitivity for drought and significant increase of expression levels under drought stress. MaSWEET17a-d showed a negative response to temperature change with a significant decrease of expression levels under low temperature and high temperature treatments (Fig. 7o−q).
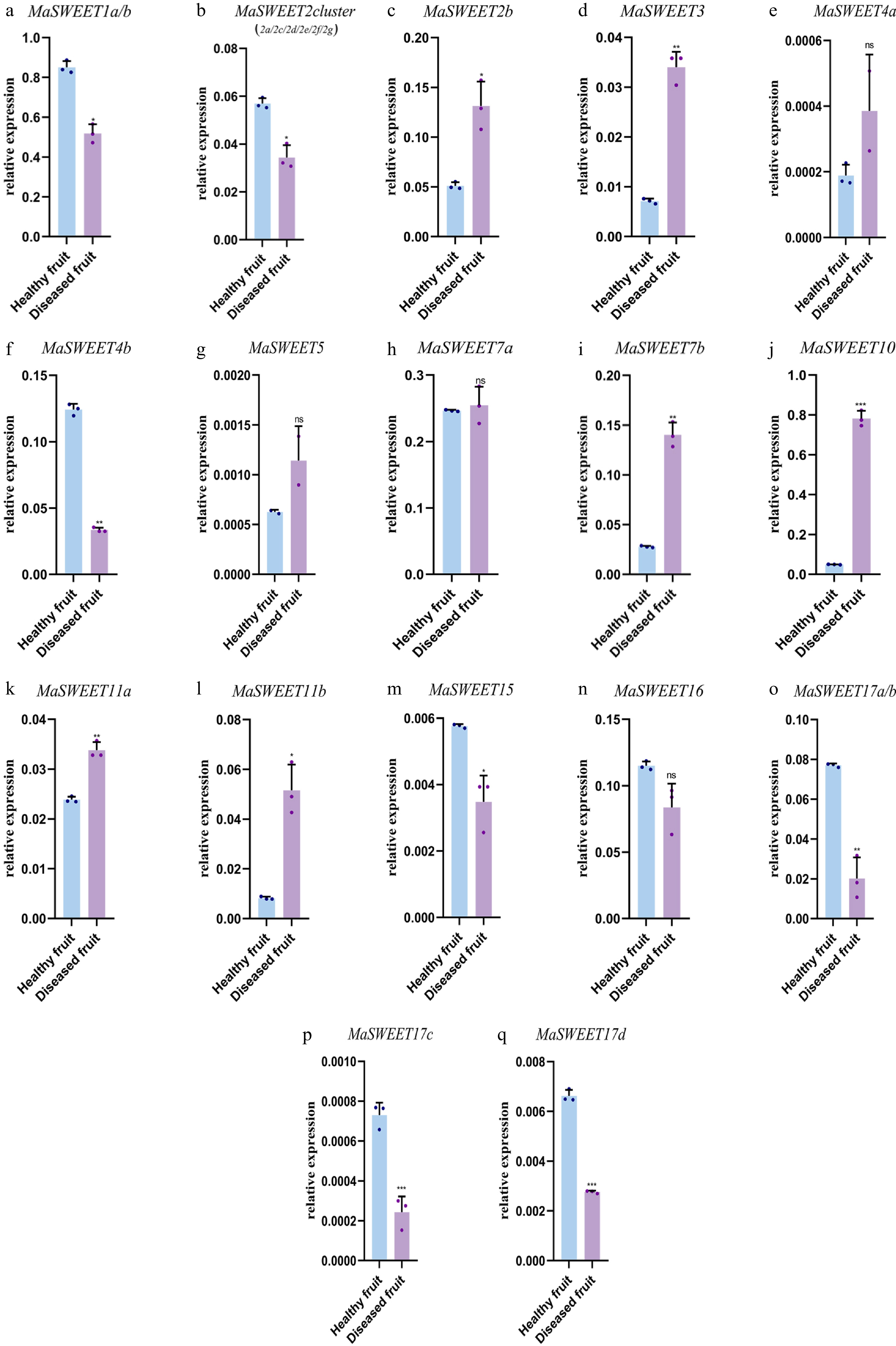
Figure 6.
Expression levels of 24 selected MaSWEET genes in response to fungi stress conditions. Three technical replicates were analyzed. Error bars represent SE. Asterisks indicate significant difference as determined by Student’s t-test (* p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001).
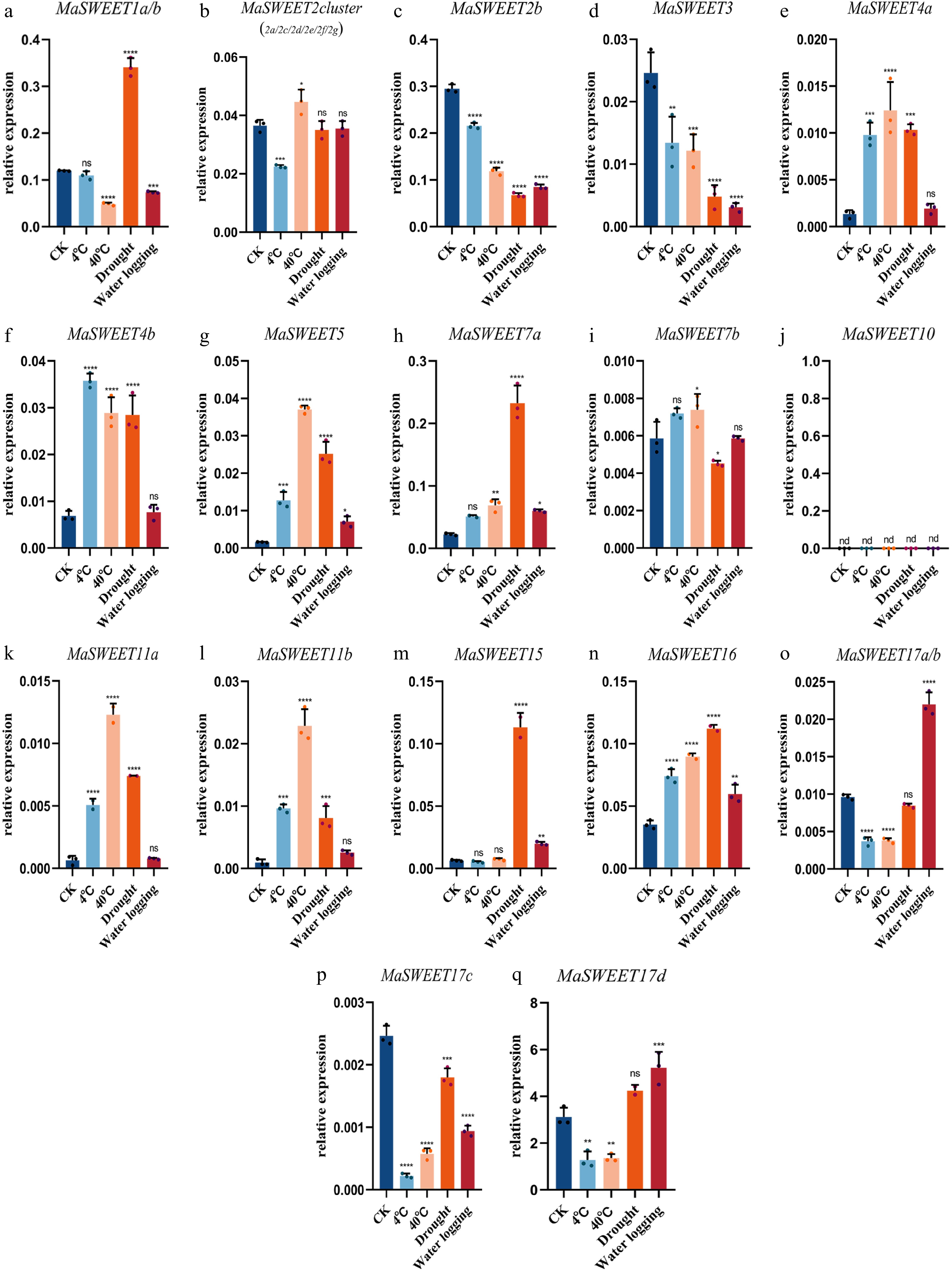
Figure 7.
Expression levels of 24 selected MaSWEET genes in response to 4 °C, 40 °C , drought, and flood stress conditions. Three technical replicates were analyzed. Error bars represent SE. Asterisks indicate significant difference as determined by Student’s t-test (* p < 0.05 ; ** p < 0.01 ; *** p < 0.001 ; **** p < 0.0001 ).
Functional characterization of MaSWEET1a in response to sclerotiniose infection
-
MaSWEET1a/b is quite different from other MaSWEETs in clade I based on expression profile analysis, which showed preferential expression in fruits and a negative response to sclerotiniose pathogen (Ciboria shiraiana) infection. Our unpublished data indicated MaSWEET1a as key genes involved in the pathogen infection process based on comparative transcriptome analysis. Transient overexpression of MaSWEET1a in tobacco and VIGS knock-down of MaSWEET1a/b in mulberry were performed. RT-qPCR results validated the successful overexpression of MaSWEET1a in tobacco and knock-down of MaSWEET1a/b in mulberry (Fig. 8b, d). The expression level of MaSWEET1a affected the resistance to C. shiraiana infection in both tobacco and mulberry (Fig. 8a, d). Overexpression of MaSWEET1a decreased the resistance to C. shiraiana infection with more severe cell death symptoms observed in OE-line tobacco (Fig. 8a). Knock-down of MaSWEET1a/b in mulberry could increase the resistance to C. shiraiana infection in mulberry (Fig. 8d). These results proved that MaSWEET1a is an important negative regulator of resistance to C. shiraiana infection in mulberry.
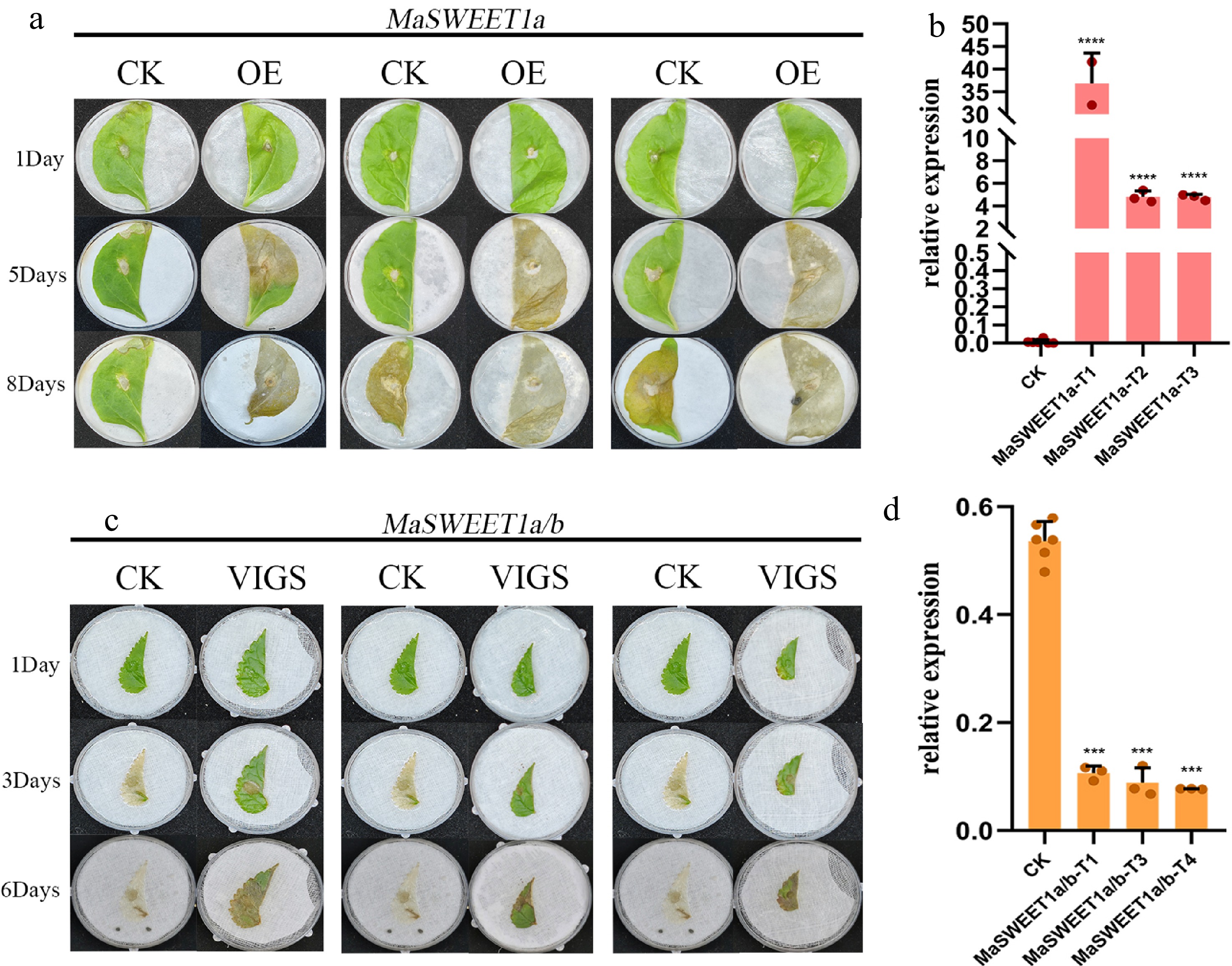
Figure 8.
Functional characterization of MaSWEET1a/b in tobacco and mulberry. (a) Damage of tobacco overexpressing MaSWEET1a after infection by C. shiraiana. (b) Expression levels of MaSWEET1a in tobacco. MaSWEET1a-T1, T2, T3 are three independent treatments with transient overexpressed MaSWEET1a. (c) Damage of mulberry with knock-down MaSWEET1a/b after infection by C. shiraiana. (d) The expression level of MaSWEET1a/b in mulberry. MaSWEET1a/b-T1, T3, T4 are three independent treatments with down-regulation of MaSWEET1a/b using VIGS.
-
Functional studies on SWEETs have revealed that SWEET homologs not only act as loading and unloading transporters of sugars but also play critical roles in various biological processes. Typically, angiosperm genomes contain about 15–25 SWEET genes[4]. In the present study, a total of 24 SWEET genes were identified and clustered into four clades corresponding with the knowledge of SWEETs in angiosperm. Several SWEETs including MaSWEET1a/b, MaSWEET4a-b, MaSWEET10, MaSWEET11a-b and MaSWEET15 showed preferential expression in fruits indicating their possible roles in fruit development. Similar expression preference of AtSWEETs was also reported in Arabidopsis[2]. It is noted that fruit-preferential expressed MaSWEETs still showed temporal expression difference during fruit development indicating time-course regulation of MaSWEETs for fruit ripening in mulberry. Early-stage expressed MaSWEET1a/b and MaSWEET4a/b further differed from late-stage expressed MaSWEET10, MaSWEET11a/b and MaSWEET15 in terms of detailed expression patterns during fruit ripening. MaSWEET2b and 2 cluster genes (MaSWEET2a, c-g) showed preferential expression in leaves which is similar with the ortholog ZjSWEET2.2 in Ziziphus jujuba. ZjSWEET2.2 was reported to be involved in mediating sugar loading in leaves[46]. MaSWEET3, 7a-b,16 and 17a-d also showed highest expression levels in leaves indicating their possible roles in sugar source loading or unloading. MaSWEET11b showed higher expression levels in phloem and its ortholog AtSWEET11 in Arabidopsis was reported to be involved in sugar phloem loading[3].
Sugar signal is critical for plants in response to various stresses. Previous studies have shown that SWEETs participated in abiotic and biotic responses in many plant species including arabidopsis and rice[21, 25]. AtSWEET11, 12, 15 and 16 were reported to be involved in affecting cold tolerance in Arabidopsis[19−21]. HfSWEET17 was also reported as a positive regulator of resistance to cold stress in daylily[11]. Cold environment (4 °C) induced expression of MaSWEET4a, 4b, 5, 11a, 11b and 16 in mulberry. Interestingly, these cold-induced MaSWEETs can also be induced by high temperature. MaSWEET15 which is the ortholog of AtSWEET15 can be induced by drought as well as low or high temperature. MaSWEET15, MaSWEET1a/b. 4a, 4b, 5, 7a, 11a, 11b, and 16 also showed positive responses to drought. It is obvious that some SWEET genes can be induced by different stresses. AtSWEET15 was also reported to be induced by osmotic, drought and salinity[21]. MaSWEET4a, 4b, 11a, 11b and 16 can be induced by low or high temperature and drought indicating their important roles in response to various abiotic stresses in mulberry.
SWEETs were generally thought to 'support the enemy' during infection. SWEETs especially those that function as exporters generally facilitate the export of sugars out of host cells, which support pathogen growth in the apoplasm[15,47,48]. Clade III SWEETs including AtSWEET11, 12, OsSWEET11 were characterized as negative regulators of resistance to fungal infection and Clade III SWEETs including OsSWEET11, 13, 14 and GhSWEET10 were characterized as negative regulators of resistance to bacterial pathogen infection[2,24,49,50]. In contrast, clade I AtSWEET2, a glucose importer and clade III IbSWEET10 were reported as positive regulators of resistance to fungal infection[48,51]. Therefore, roles of SWEETs in response to pathogen infection may be quite different. Most MaSWEETs were disturbed in diseased fruits that resulted from sclerotiniose pathogen infection. MaSWEET2b, MaSWEET3 in clade I, MaSWEET7b in clade II, MaSWEET10 and MaSWEET11a-b in clade III showed significant increase in expression levels in diseased fruits while MaSWEET1a/b, MaSWEET2 cluster (MaSWEET2a, c-g) in clade I showed significant decrease of expression levels in diseased fruits. MaSWEET1a was further validated as a negative regulator of resistance to C. shiraiana infection. Given the fact that MaSWEET1a/b was repressed in diseased fruits, it is likely that a possible pathway through repression of MaSWEET1a exists in mulberry to defense pathogen infection.
In conclusion, we have performed a genome-wide investigation of SWEET genes in Morus and a comprehensive analysis including phylogenetic analysis, promoter analysis and expression profile analysis was also carried out. Their possible roles in development and response to abiotic and biotic stresses were addressed. In particular, the functional role of MaSWEET1a in regulation of tolerance to C. shiraiana infection was validated using both VIGS knock-down and transient overexpression in tobacco combined with inoculation of C. shiraiana. The results in this study provides a foundation for studying the function of the SWEET family in mulberry plants and provides a negative regulator of resistance to C. shiraiana infection for further genetic modification.
This work was jointly supported by the National Natural Science Foundation of China (32201526), Crop Germplasm Resources Protection Project of the Ministry of Agriculture and Rural Affairs of the People's Republic of China (19200382), National Infrastructure for Crop Germplasm Resources (NCGRC-2020-041), and China Agriculture Research System of MOF and MARA (CARS-18). We thank Professor Aichun Zhao at Southwest University for providing us C. shiraiana strains and Professor Feng Jiao at Northwest University of Agriculture and Forestry who provided us the genome annotation file of M. alba.
-
The authors declare that they have no conflict of interest.
-
# These authors contributed equally: Xiaoru Kang, Shuai Huang, Yuming Feng
- Supplemental Table S1 Primers used in this study.
- Supplemental Table S2 Putative cis-elements of MaSWEETs promoters.
- Supplemental Fig. S1 Prediction of MaSWEETs transmbrane region.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Kang X, Huang S, Feng Y, Fu R, Tang F, et al. 2023. SWEET transporters and their potential roles in response to abiotic and biotic stresses in mulberry. Beverage Plant Research 3:6 doi: 10.48130/BPR-2023-0006 

SWEET transporters and their potential roles in response to abiotic and biotic stresses in mulberry
- Received: 24 November 2022
- Revised: 07 January 2023
- Accepted: 17 January 2023
- Published online: 02 March 2023
Abstract: Mulberry (Morus spp., Moraceae) is a traditional economic crop plant and is also being gradually utilized as a beverage plant. SWEETs (Sugars Will Eventually be Exported Transporter) are important sugar transporters involved in various biological processes and responses to various stresses. However, SWEETs in mulberry are still poorly studied without a comprehensive functional analysis of SWEETs. In the present study, a total of 24 SWEETs were identified using the Morus alba (Ma) genome. Phylogenetic analysis showed that these 24 MaSWEETs were clustered with SWEETs from Arabidopsis, Populus and Oryza and fell into four clades. MaSWEETs in the same clade are likely to pose similar intron/exon patterns. These MaSWEETs distributed on 12 chromosomes and tandem duplication and block duplication were responsible for the expansion of SWEETs in mulberry. Transmembrane domains and conserved active sites of Tyr and Asp were observed in MaSWEETs. Cis-elements in promoter regions of MaSWEETs indicated the possible function of MaSWEETs in response to hormones and environment stimulus. MaSWEETs showed quite different expression preference in tissues and organs indicating the possible function divergence. In addition, most MaSWEETs showed a disturbed expression levels in response to various abiotic stresses and Ciboria shiraiana infection. MaSWEET1a was functionally characterized as a negative regulator of resistance to C. shiraiana infection based on in vivo transient overexpression of MaSWEET1a in tobacco and down-regulation of MaSWEET1a/b in mulberry. Our results provided foundation for further functional dissection of SWEETs in mulberry and a potential regulator for genetic modification.
-
Key words:
- Mulberry /
- SWEET /
- Abiotic stress /
- Fungal pathogen /
- Functional divergence
{kind=link}