









-
Carboxypeptidase A (CPA) (EC 3.4.17.1) is a metallocarboxypeptidase belonging to the M14 family. It consists of 307 amino acids with a molecular weight of 34.472 kDa, which are mainly divided into two groups, CPA1 and CPA2. As an exopeptidase, CPA exclusively releases amino acids from the C-terminal region of peptides or proteins[1]. CPA1 hydrolyzes smaller aliphatic amino acids and CPA2 prefers to hydrolyze larger aromatic amino acids. In the structure of CPA, there is a Zn2+ essential for the enzyme activity[2]. It was found that CPA has significant applications in industrial fields. Firstly, CPA with excellent ochratoxin A (OTA) degradation activity hydrolyzes toxic OTA into non-toxic ochratoxin α (OTα) and L-phenylalanine[3], showing great potential for OTA degradation in the food and feed industry. In addition, CPA can not only remove the bitterness of peptides in food industries and reduce food allergies, but also be applied in manufacturing functional or fermented foods[4]. As a mesophilic enzyme, the catalytic activity of CPA tends to decrease rapidly or inactivate at elevated temperatures, seriously limiting its industrial application.
High thermostability is always considered to be a crucial prerequisite to evaluating the feasibility of enzymes for industrial applications[5]. Typically, higher thermostability confers greater competitiveness and wider applicability to enzymes[6]. Enzymes with thermostability can maintain long-lasting catalytic activity reducing the reaction time and the production cost. However, most natural enzymes perform their catalytic function under relatively mild conditions. They are not well adapted to harsh industrial processes. Therefore, adopting appropriate strategies to improve the thermostability of biological enzymes is of great value for industrial and biomedical applications. Protein engineering, as one of the most ideal strategies to improve enzyme thermostability, mainly includes three approaches (rational design, directed evolution, and semi-rational design)[7]. Rational design is based on an understanding of the relationships between enzyme structure and function[8]. It can quickly predict target regions and modification sites with the help of computer programs. It has the advantages of a small screening workload, high prediction accuracy, and efficiency. Currently, rational design strategies successfully applied include homology comparison[9], introduction of salt bridges[10] or disulfide bonds[11], substitution of proline[12] or glycine[13]. In particular, the disulfide bond is formed between the thiol groups of two spatially close cysteine residues, which is significant for enzyme folding, stability, and function[14]. As a covalent interaction, it maintains stability of the structure of enzymes by reducing conformational entropy. It was found that the energy provided by disulfide bonds to maintain enzyme stability was greatest compared with other non-covalent interaction forces such as hydrogen bonds and salt bridges[15]. In recent years, a large number of studies have shown that the rational design of introducing disulfide bonds could realize the improvement of enzyme thermostability. For example, Zhang et al. introduced a disulfide bond at the C-terminus of B. licheniformis phytase, and successfully improved the thermostability of it[16]. In addition, transglutaminase[17], lipase[18], and ammonia lyase[19] also achieved enhanced thermostability by rational design of disulfide bonds. However, not all disulfide bond introductions were beneficial. The introduction of disulfide bonds at the N-terminus negatively affected the thermostability of the 1,3-1,4-β-glucanase mutant V59C-Y86C[20]. Therefore, it is crucial to select appropriate strategies for introducing disulfide bonds in suitable regions or sites. A previous study showed that strategically introducing disulfide bonds in flexible regions could provide stronger rigid support and create a protected microenvironment for the enzyme, effectively improving thermostability and catalytic efficiency[15].
In our previous study, the thermostability of CPA was successfully enhanced by multiple computer-aided rational design based on amino acids preferences at β-turns while the catalytic activity was significantly decreased[21]. In this study, the rational design of disulfide bonds by HotSpot Wizard program and molecular dynamics (MD) simulations was proposed to improve the thermostability of CPA and reduce the loss of activity in ochratoxin degradation by obtaining mutants and studying the molecular mechanism responsible for thermostability.
-
The strains E. coli DH5α carrying pPIC9K plasmids and P. pastoris GS115 were stored at our lab. The restriction enzyme Sac I was purchased from Takara Biotechnology Co., Ltd. (Beijing, China). The SDS-PAGE fast protein gel kit was purchased from Dakewe Biotechnology Co., Ltd. (Beijing, China). Detergent Compatible Bradford Protein Assay Kit was purchased from Beyotime Biotechnology Co., Ltd. (Shanghai, China). HiTrapTM Phenyl FF prepacked column was purchased from GE HealthCare (London, UK). The composition of liquid yeast extract peptone dextrose (YPD) medium was 10 g·L−1 yeast extract, 20 g·L−1 tryptone, and 20 g·L−1 glucose. The composition of solid minimal dextrose (MD) medium was 13.4 g·L−1 yeast nitrogen base without amino acids (YNB), 20 g·L−1 glucose, 400 μg·L−1 D-biotin and 15 g·L−1 agar. All other chemicals were analytical or chromatographic grade and were purchased from China National Pharmaceutical Group Corporation (Shanghai, China).
Rational design of disulfide bonds of CPA
-
The crystal structure of Bos taurus CPA (PDB ID:1M4L) was derived from the PDB database (
www.rcsb.org ). The three-dimensional structures of mutants were obtained through SWISS-MODEL online server using CPA crystal structures as templates. The quality of 3D models of mutants was evaluated by ERRAT and PROCHECK procedures in SAVES v6.0 online server (https://saves.mbi.ucla.edu/ ). The structures above the lowest overall quality factor of 90 in REEAT and more than 95% allowed area in Ramachandran plots were used for further analysis[22]. B-Factors of CPA were predicted by HotSpot Wizard 3.0 online procedure (https://loschmidt.chemi.muni.cz/hotspotwizard/ ). Potential residue pairs for introducing disulfide bonds were synthetically analyzed through Disulfide by Design 2.0 online server (http://cptweb.cpt.wayne.edu/DbD2/index.php ) and MODIP online procedure (http://caps.ncbs.res.in/dsdbase2 ). The position-specific scoring matrix (PSSM) of CPA was generated by the PSI-BLAST algorithm. MD simulations were performed by the Gromacs software following a previously described method by Ming et al. with appropriate modifications[21]. Firstly, it was conducted by Amber99sb-Idn force filed at 370 K for 20 ns. Then, the 3D model of WT and its mutants was wrapped with a water molecule box, respectively. Then, Na+ ions were added to neutralize the charges of the system. The energy minimization using the steepest descent method was subjected to reduce the number of modeled non-normal conformations in the simulated system. Furthermore, a 500-ps isothermal isotropic ensemble (NVT) and isothermal isobaric ensemble (NPT) were executed to balance the entire system, respectively. The RMSF values of WT were calculated to determine the flexible regions of CPA, where residues with RMSF higher than 0.145 nm were considered highly flexible. The Root-Mean-Square Deviation (RMSD) and the root-mean-square fluctuation (RMSF) values of WT and its mutants were determined to assess changes in CPA conformation.Construction of recombinant plasmids and strains
-
The gene sequence of CPA and its mutants (D93C/F96C and K153C/S251C) were optimized based on the codon preference of P. pastoris. Subsequently, they were inserted into the EcoR I and Not I restriction sites of pPIC9K plasmids, respectively. The pPIC9K plasmids containing the cpa gene were stored in E. coli DH5α strains. The above process was constructed by Sangon Biotech Co., Ltd. (Shanghai, China). These recombinant plasmids linearized by Sac I were transferred into P. pastoris competent cells by electrotransformation. The products were successively cultured at 28 °C in histidine-deficient solid minimal dextrose (MD) plates for screening His+ clones. Then, recombinants with different copy numbers of genes were screened by solid YPD mediums containing geneticin G418 at a final concentration of 0.5, 1.0, 2.0, and 4.0 mg·mL−1.
Expression and purification
-
The positive clones of CPA and its mutants were cultured at 28 °C, 200 rpm in 25 mL liquid BMGY medium until the OD600 reached 2~6. After the centrifugation at 4,000 rpm for 5 min, the cells were transferred in 20 mL liquid BMGY medium and cultured at 28 °C, 250 rpm for 6 d. The methanol was added to a final concentration of 1% (v/v) every 24 h to induce protein expression. After the centrifugation at 10,000 rpm for 5 min, crude supernatants were analyzed by SDS-PAGE and purified by hydrophobic interaction chromatography. The HiTrapTM Phenyl FF prepacked column was pre-equilibrated in 2 M (NH4)2SO4 (pH 7.4). After fully loaded, the enzymes treated with 2 M (NH4)2SO4 were eluted with 20 mM Tris-HCl (pH 7.4). The SDS-PAGE and Western-Blot analysis of pure enzymes were performed following a previous method described by Ming et al.[21]. And, the concentration of pure enzyme was measured by the Bradford method while using bovine serum albumin (BSA) as the standard.
Activity assays
-
The determination of OTA degradation activity of recombinant CPA and its mutants were detected by HPLC, which was described previously[23]. The activity of recombinant CPA and its mutants was determined using Z-Phe-Leu as the substrate. One unit (U) of activity was defined as the amount of enzyme required to degrade 1 μM Z-Phe-Leu per minute at 37 °C. The reaction system contained 108 μL of Z-Phe-Leu (1 μM ) and 12 μL of purified CPA (10 mg·mL−1), and incubated at 37 °C for 15 min. Then, 240 μL of cadmium-ninhydrin solution was added to it and treated at 85 °C for 5 min. The absorbance of samples at 507 nm was recorded in a spectrophotometer.
Thermal activity assays
-
To determine the optimum temperature (Topt) of recombinant CPA and its mutants, the reaction system contained Z-Phe-Leu, and purified enzymes were incubated at various temperatures ranging from 30 to 70 °C with 10 °C intervals for 15 min. The residual activity was assayed at 30 °C, and the activity at the optimum temperature was considered as 100%.
To determine the half-time (t1/2) of recombinant CPA and its mutants, the purified enzymes were heat-treated at 65 °C for 10, 20, 30, 40, and 50 min, and then cooled on ice for 10 min. The residual activity was assayed at 30 °C with the initial activity set at 100%. The average thermal inactivation rate constants (Kd) were obtained from the plots of ln (residual activity) vs the incubation time. The value of t1/2 is defined as the time when the residual activity is reduced to half, and it can be calculated by the equation: t1/2 =(ln2/Kd).
To determine the half-inactivation temperature (T5015) of recombinant CPA and its mutants, the purified enzymes were incubated at temperatures ranging from 40 to 70 °C with 10 °C intervals for 15 min, and then cooled on ice for 10 min. The residual activity was assayed at 30 °C with the initial activity set at 100%. The value of T5015 is the temperature at which the residual activity is decreased to 50% after a 15-min treatment, and it can be generated from the plot of residual activity versus the incubation temperature.
Kinetic parameters analysis
-
The kinetic parameters of purified CPA and its mutants were determined based on the enzyme activity assays. Different final concentrations of Z-Phe-Leu (0.5~2.5 μM) were incubated with CPA at 37 °C for 15 min, respectively. Then, the kinetic parameters of Km and Kcat/Km were calculated by the Line Lineweaver–Burk plots in Origin 2021 software (Origin Lab, USA).
Advanced structures analysis
-
The circular dichroism (CD) spectra were conducted on a Pistar II-180 circular dichroism spectrometer (Applied Photophysics, USK). The purified CPA and its mutants prepared in 20 mg·mL−1 Tris−HCl buffer (pH 7.4) were added to the length quartz cuvette with a path length of 0.1 cm. The wavelength range was set from 200 to 260 nm, and the scan speed was 10 nm·s−1. Each scan was repeated three times. The proportion of secondary structures (α-helix, β-sheet, β-turns, and coils) were estimated by CDPro online software (
www.bmb.colostate.edu/cdpro ).The intrinsic fluorescence spectra were performed on a Hitachi F-7000 fluorescence spectrometer (Hitachi, Japan) to evaluate the tertiary structure changes. The purified CPA and its mutants prepared in 20 mmol·L−1 Tris−HCl buffer (pH 7.4) were added to the length quartz cuvette with a path length of 0.1 cm. The excitation wavelength was at 280 nm, and the emission wavelength was recorded ranging from 290 to 500 nm.
Intramolecular interactions analysis
-
The disulfide bonds of recombinant CPA and its mutants were quantified mainly according to the content of the free sulfhydryl group when in reduction. Firstly, reduced enzymes were prepared in the following way. They were firstly dissolved in 2 mL disulfide bond reduction buffer and treated at 37 °C for 2 h. Subsequently, 50 μL of TCEP was added to the solution and then treated at 37 °C for 1 h. The solution was to added 2 mL 12% trichloroacetic acid (TCA) and treated at 37 °C for 1 h. After centrifugation at 14,000 g for 10 min, the precipitate was dissolved with 2 mL Tris-Gly buffer. Non-reduced recombinant CPA and its mutants were dissolved in 2 mL Tris-Gly buffer. Fifteen μL of DNTB (5,5'-disulfide (2-nitrobenzene) was added to 1 mL reduced and non-reduced recombinant enzymes, respectively. After the reaction solution was placed at 37 °C for 20 min, the wavelength of 412 nm was detected by ultraviolet spectrophotometer. Disulfide bonds were calculated by the equation: n = N × (A1 − A2)/2 × A1. n was the number of disulfide bonds, N was the number of cysteine residues, A1 was the absorption value of reduced recombination CPA at 412 nm, A2 was the absorption value at 412 nm for non-reduced recombination CPA.
The ProteinTools online server (
https://proteintools.uni-bayreuth.de ) was employed to analyze changes in hydrogen bonds between WT and its mutants. PyMOL v2.5 was conducted to visualize alterations in surface charge.Statistical analysis
-
All results were expressed as mean value ± standard deviation based on three independent experiments. And, figures were drawn using Origin 2021 software.
-
The enzyme flexible regions with high conformational flexibility are always used as a potential target for molecular modification in protein engineering. The flexible regions of CPA were mainly targeted by two methods (B-factor and RMSF). The B-factor is used to characterize the describe the attenuation of X-ray or neutron scattering caused by thermal motion. Where the higher the B-Factor value, the more flexible the enzyme conformation is[24]. As seen in Fig. 1, the average B-Factor value of residue was calculated by HotSpot Wizard 3.0, and a total of 16 highly flexible regions of CPA were identified.
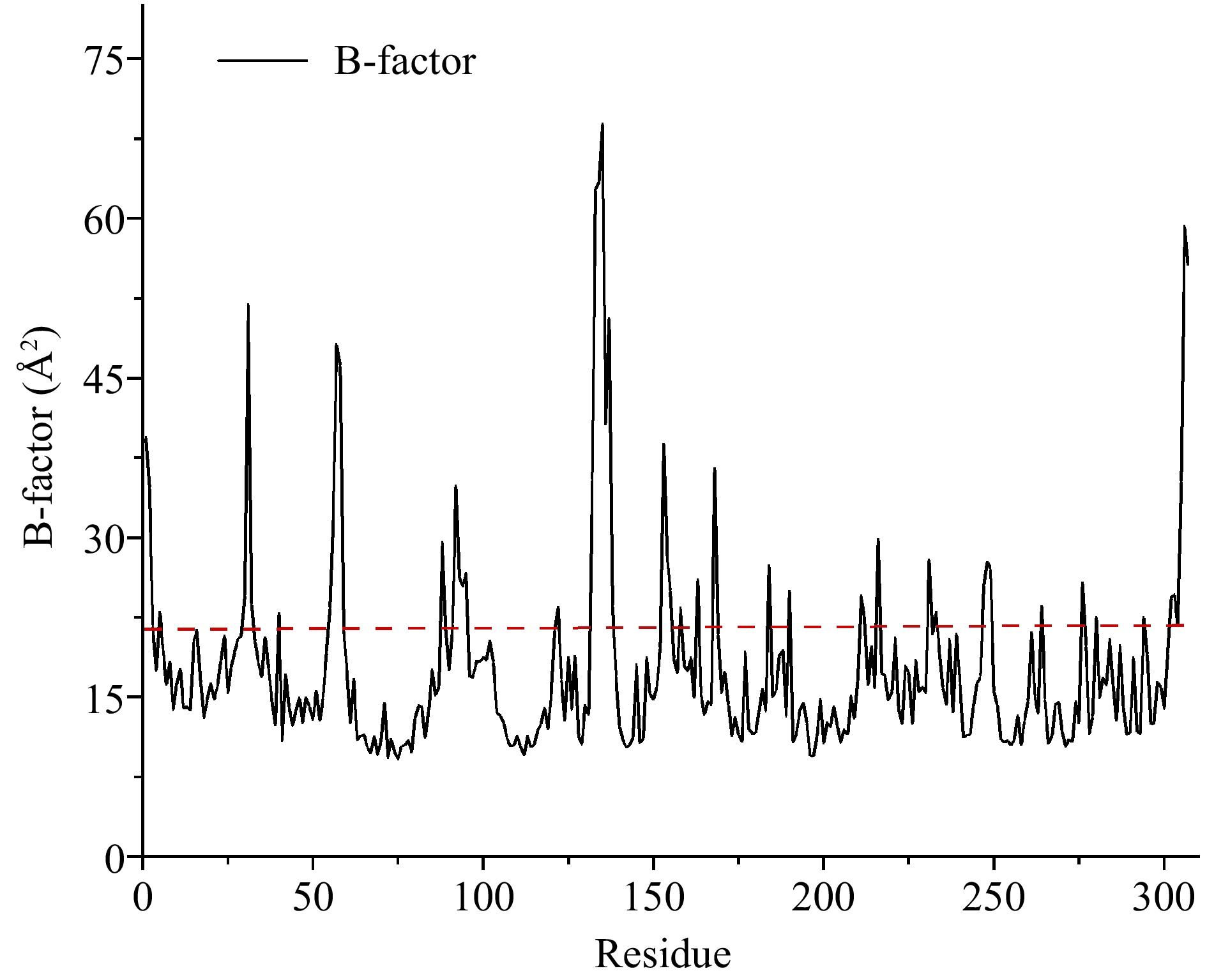
Figure 1.
Average B-factor values of carboxypeptidase A. Flexible residues were located above the dotted line.
RMSF represents the amplitude of fluctuation of an atom relative to its average position in the simulated system, reflecting the local flexibility of protein conformation at the atomic level[21]. The higher the RMSF value, the more flexible the enzyme conformation is. As shown in Fig. 2, the RMSF values of CPA were calculated by molecular dynamics simulation, and a total of twelve flexible regions of CPA were screened.
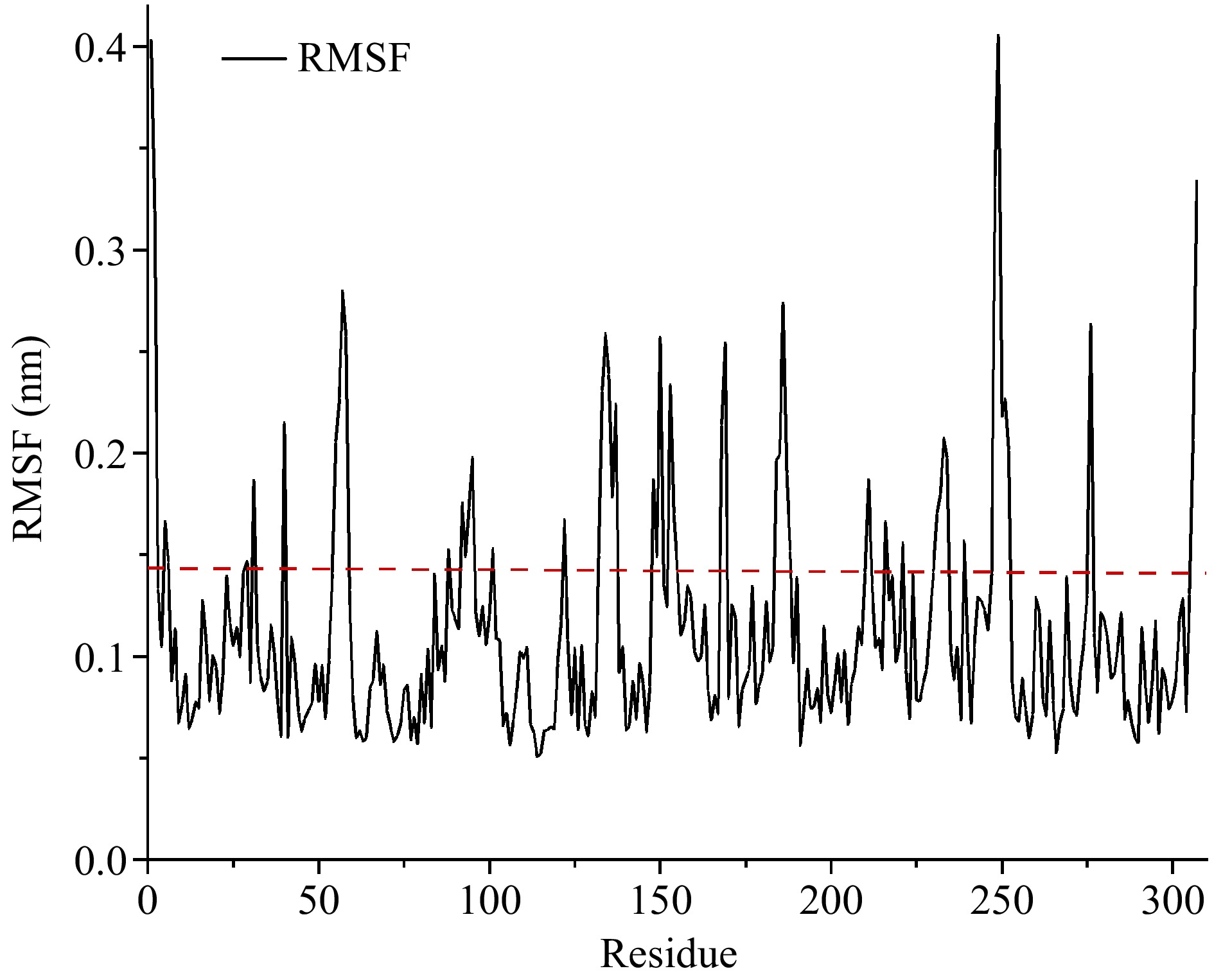
Figure 2.
The RMSF values of carboxypeptidase A. Flexible residues were located above the dotted line.
To improve the prediction accuracy, B-factor and RMSF values were comprehensively analyzed. Finally, the regions of Ala1-Arg2, Asn5-Thr6, Gly55-Asn58, Gln92-Ser95, Val132-Leu137, Lys153-Ala154, Lys168-Tyr169, Lys231-Tyr234, Try248-Gln249, and Asn306-Asn307 were considered highly flexible regions, laying the foundation for the subsequent rigidification of flexible regions.
Selection of potential disulfide bonds in flexible regions of CPA
-
The introduction of disulfide bonds is always one of the strategies to stabilize the flexible regions. Potential residue pairs for introducing disulfide bonds in CPA were analyzed by two online programs, DbD and MODIP (Supplemental Fig. S1). There were 37 pairs of potential disulfide bonds in CPA predicted by DbD software. Besides, there were eight pairs of potential disulfide bonds predicted by the MODIP program, and they were categorized into Grade A, Grade B, Grade C and Grade D according to the likelihood of disulfide bond formation. To improve the prediction reliability of potential disulfide bonds in CPA, twenty potential disulfide bonds belonging to both DbD and MODIP classes A and B were screened. The residues including Asp93, Ser136 and Lys153 were located in these flexible regions. Therefore, three pairs of residues for introducing disulfide bonds Asp93-Phe96, Ser136-Pro160 and Lys153-Ser251 were initially screened for subsequent analysis.
Analysis of conserved residues of CPA
-
Enzymes not only need enough rigidity to maintain their stability, but also require adequate conformational flexibility to exert its catalytic activity. Thus, there is a trade-off between activity and stability, implying that an increase in stability is accompanied by a concomitant decrease in activity[25]. It was reported that residues associated with the catalytic function of enzymes were typically highly conserved[26]. A total of 151 conserved residues in the CPA sequence were determined by conservativeness analysis of PSSM, containing the screened residue Ser136 (Supplemental Fig. S2). Thus, the disulfide bond Ser136-Pro160 was excluded to prevent the catalytic activity of CPA from being adversely affected when engineering thermostability. Finally, the two CPA disulfide bonds Asp93-Phe96 and Lys153-Ser251 were retained and sequentially named as mutants D93C/F96C and K153C/S251C.
Homology Modeling of CPA mutants
-
Using the crystal structures of CPA (PDB ID:1M4L) as a template, three-dimensional structures of mutants D93C/F96C and K153C/S251C were obtained through SWISS-MODEL online server. The quality of the models was evaluated through the SAVES online platform. The results of Ramachandran plots showed that mutants D93C/F96C and K153C/S251C had more than 95% residues in the favorable regions (Supplemental Fig. S3). And, the results of ERRAT indicated that the overall quality factors of D93C/F96C and K153C/S251C were 95.2703 and 95.5932, respectively (Supplemental Fig. S4). Thus, the three-dimensional models of these two mutants were relatively reliable and closer to their real structures.
Evaluation of CPA mutants with potential enhanced thermostability
-
MD simulations were performed to explore the conformational stability of CPA structure. RMSD reflects the average amount of movement of backbone atoms throughout the whole protein structure, which is negatively correlated with the thermostability of the enzyme[21]. The average RMSD values of mutants D93C/F96C and K153C/S251C were 0.1386 nm (Fig. 3a) and 0.1136 nm (Fig. 3b), which were lower than that of WT (0.1416 nm). It suggested that mutations could make the overall conformation more rigid, contributing to enhancing the thermostability of CPA[27].

Figure 3.
The comparison of RMSD values between wild-type and mutant carboxypeptidase A. (a) WT and D93C/F96C. (b) WT and K153C/S251C.
The effect of mutation on the local conformational stability of CPA was explored by RMSF. Compared with WT, the mutants D93C/F96C (Fig. 4a) and K153C/S251C (Fig. 4b) both showed a decrease in RMSF values in mutant regions, showing that the mutations stabilize the local conformation of CPA. Taken together, the two mutants D93C/F96C and K153C/S251C possessed the potential to enhance the thermostability compared with WT. And, they were used for the subsequent experimental analysis of CPA thermostability screening.
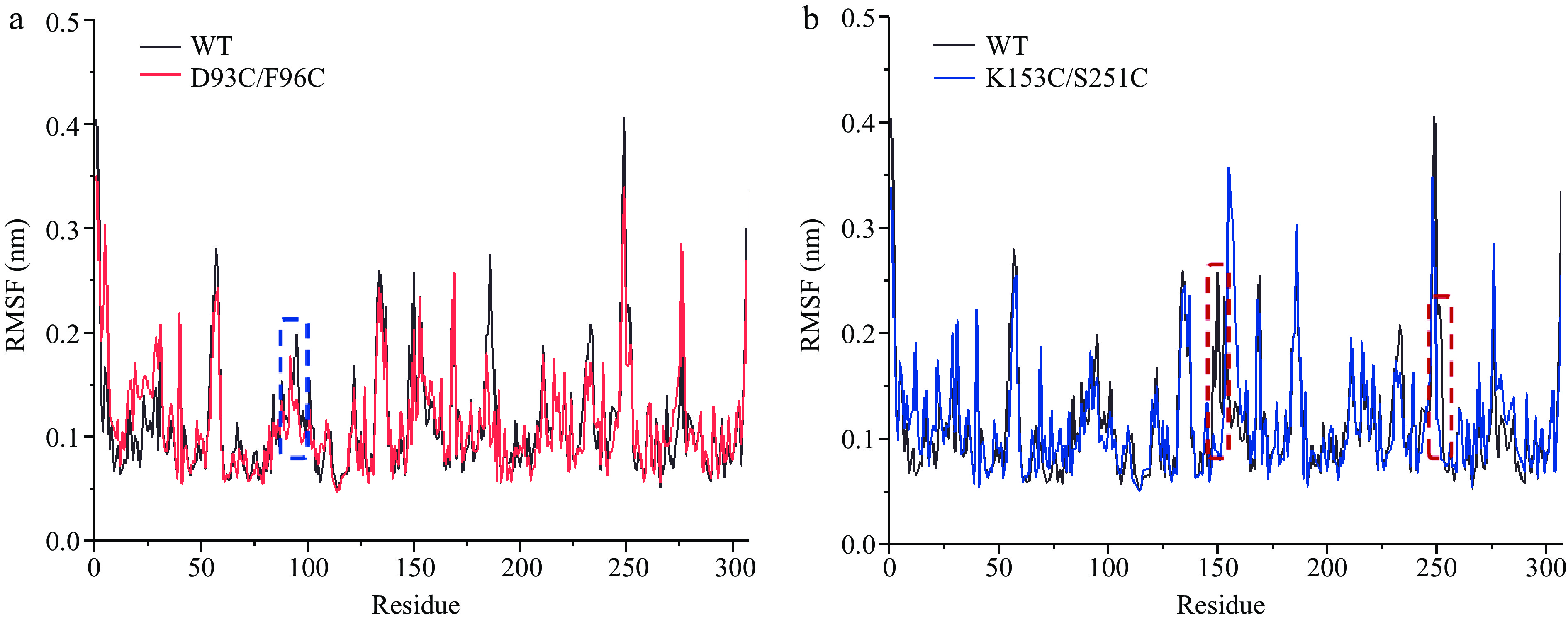
Figure 4.
The comparison of RMSF values between wild-type and mutant carboxypeptidase A. (a) WT and D93C/F96C. (b) WT and K153C/S251C. The mutant regions are circled in dotted wireframes.
Construction, expression, and purification of recombinant CPA and its mutants
-
The plasmid pPIC9K contains the histidine dehydrogenase sequence (HIS4). It was shown that a large number of single colonies capable of synthesizing histidine grew on the MD plate (Supplemental Fig. S5). The result showed that the recombinant plasmid pPIC9K had been successfully transferred into host cells.
Because the plasmid pPIC9K contains the kanamycin resistant gene that allows P. pastoris to tolerate G418 disulfate salt. The result showed that the number of viable transformants gradually decreased as the concentration of genistein increased (Supplemental Fig. S6). To some extent, the higher the level of resistance to the genotoxin G418, the higher the copy number of the target gene, which may increase the level of enzyme expression. Therefore, the transformants growing on YPD plates with 4 mg·mL−1 G418 were selected for expression.
From the SDS-PAGE result of fermentation supernatants, there was a band appeared near 35 kDa, which was more similar to the theoretical molecular weight of CPA (Fig. 5a). The result preliminarily suggested that P. pastoris GS115 realized the heterologous expression of CPA and its mutants. To improve the purity of recombinant enzymes, hydrophobic interaction chromatography was used for purification. And, the SDS-PAGE of purified WT, D93C/F96C and K153C/S251C was shown in Fig. 5b. The purity of purified enzymes exceeded 90% detected by Image J. There were six histidine tags at the N-terminal of recombinant enzymes, the specific bands in the western-blot experiment once again proved that the purified enzymes with a molecular weight of 35 kDa was recombinant CPA and its mutants (Fig. 5c). The concentration of WT, D93C/F96C and K153C/S251 measured by Bradford's method were 0.321, 0.303, and 0.289 mg·mL−1, respectively.
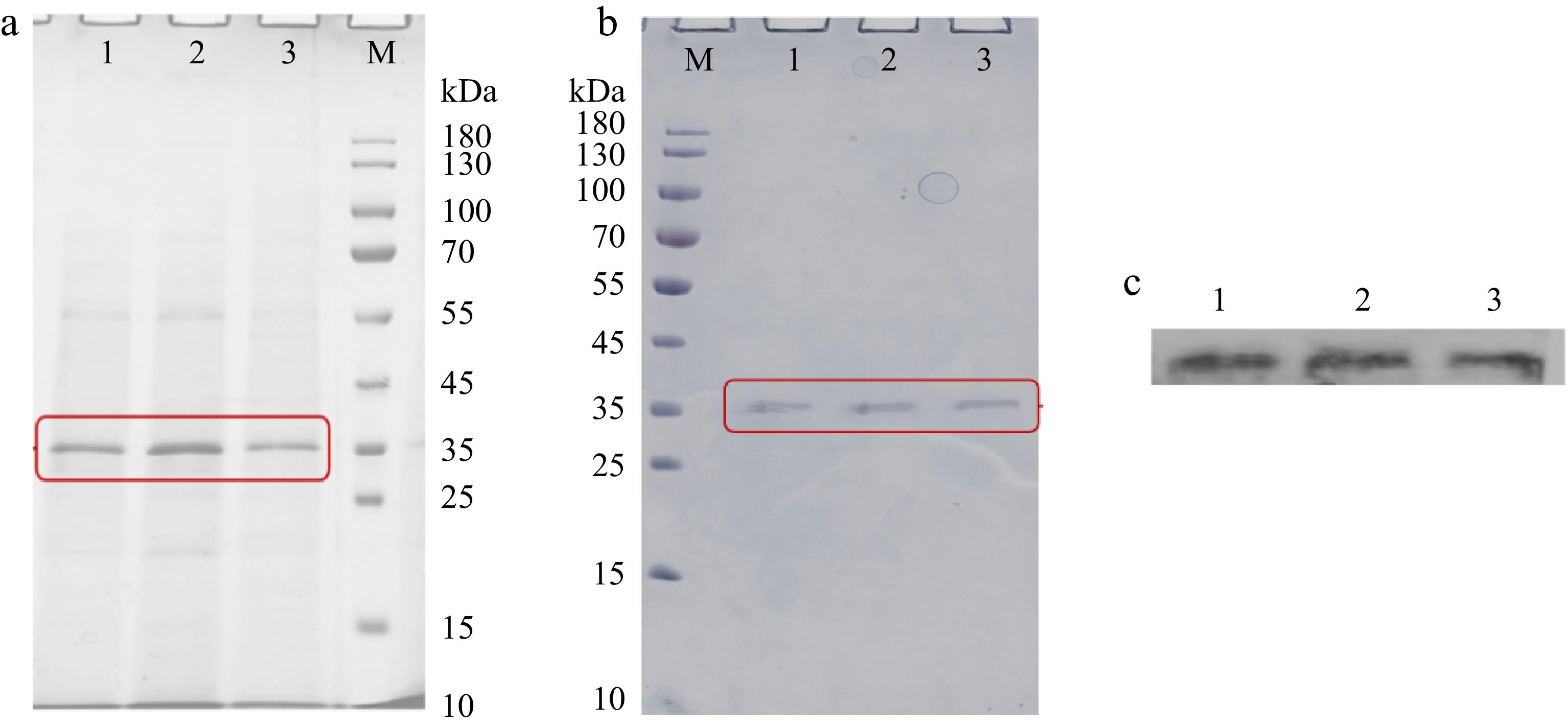
Figure 5.
The SDS-PAGE analysis of (a) fermentation supernatants and (b) purified components expressed by P. pastoris. (c) The western blot analysis of purified components expressed by P. pastoris. M, protein marker (10−180 kDa). 1, WT. 2, D93C/F96C. 3, K153C/S251C. Each sample was prepared by boiling for 5 min and loaded at 20 μL per lane.
Enzymatic analysis of recombinant CPA and its mutants
-
To determine the OTA degradation efficiency of recombinant CPA and its mutants, the residual OTA in the reaction system was detected by HPLC. It can be seen in Fig. 6 that the peak time of the OTA standard was 4.150 min, and the peak time of the OTα standard was 3.205 min. Compared with the control, the peak area of samples treated with recombinant enzymes decreased significantly at 4.1 min, indicating that OTA was successfully degraded. A new chromatographic peak appeared around 3.2 min, and its retention time was consistent with that of the OTα standard. The result indicated that the OTA degradation product of WT, D93C/F96C and K153C/S251C was OTα.
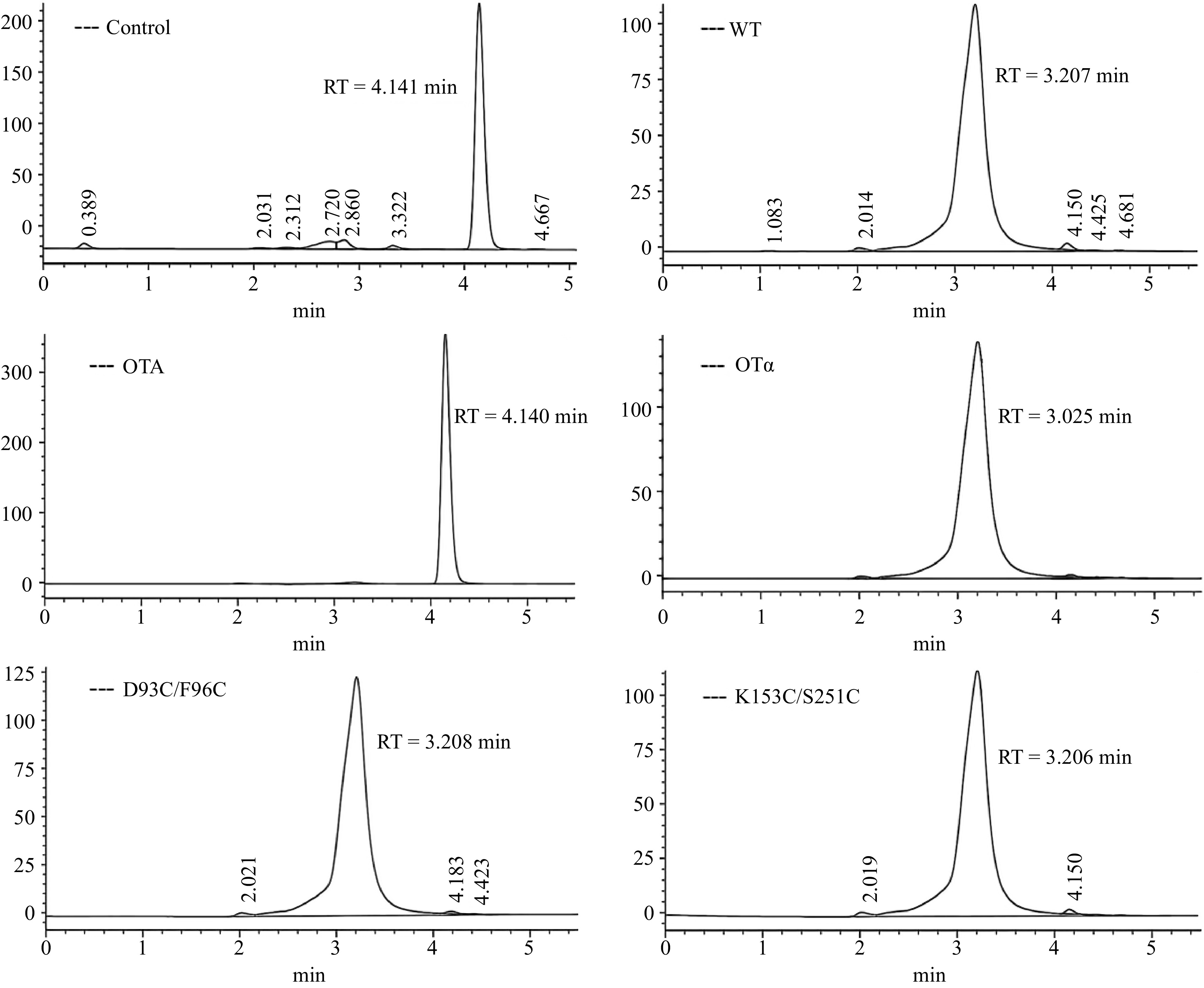
Figure 6.
HPLC of OTA degradation by recombinant CPA and its mutants.
It was found that CPA hydrolyzed the amide bond of OTA to generate OTα and L-phenylalanine[21], which was consistent with the principle of hydrolyzing Z-Phe-Leu. Given the toxicity of OTA, Z-Phe-leu was used as a substrate to study the enzymatic properties of CPA. The specific enzyme activity of recombinant WT, D93C/F96C and K153C/S251C was determined by the standard curve of L-leucine solution y = 0.0055x + 0.0857 (R2 = 0.995). The specific enzyme activity was 11.113 ± 0.298, 13.816 ± 0.511, and 10.107 ± 0.255 U·mg−1 for WT, D93C/F96C and K153C/S251C, respectively. The specific enzyme activity of D93C/F96C was increased by 24.32% compared with that of WT, indicating that the introduction of the disulfide bond at sites 93 and 96 did not adversely affect the activity. While the specific enzyme activity of the mutant K153C/S251C was reduced by 9.05% compared with that of WT, showing that the mutations at sites 153 and 251 adversely affected the enzymatic activity of CPA.
Thermostability measurements of recombinant CPA and its mutants
-
The reaction system of recombinant enzymes and substrate Z-Phe-Leu was processed at different temperatures for a certain period. And, the residual enzyme activity of recombinant enzymes was measured. As shown in Fig. 7a, the activity increased and then decreased with the increase of reaction temperature, and reached the highest at the optimal temperature. The maximum activity of WT at 40 °C indicated that the optimum temperature of WT was 40 °C. While the optimal temperatures of mutants D93C/F96C and K153/S251 were 10 °C higher than that of WT. The results showed that mutations improved the optimal temperature, which was conducive to the better catalytic activity of CPA at higher temperatures.
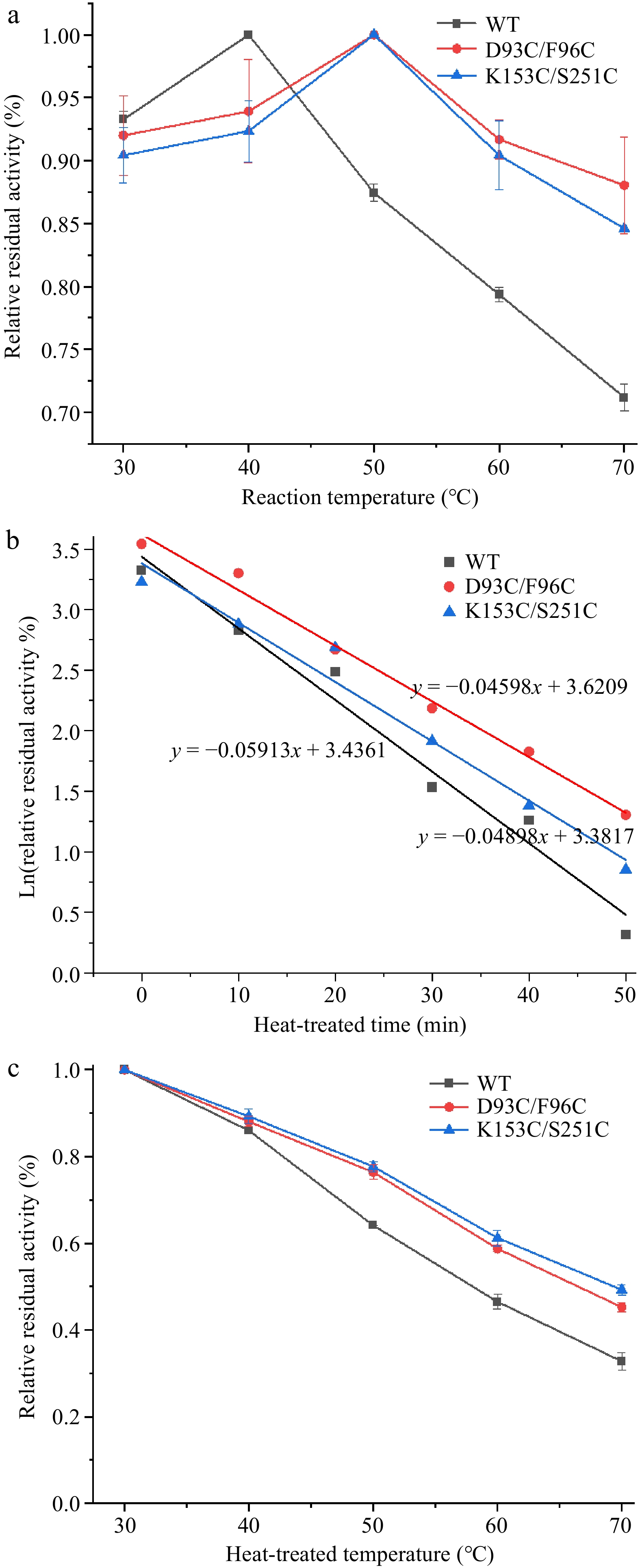
Figure 7.
Thermal stability of wild-type and mutant carboxypeptidase A. (a) The optimum temperature of wild-type and mutant carboxypeptidase A. (b) The half-life of wild-type and mutant carboxypeptidase A. (c) Half inactivation temperature of wild-type and mutant carboxypeptidase A.
The half-life (t1/2) represents the time required for the enzyme to lose half of its activity. The WT, D93C/F96C and K153C/S251C was subjected to treatment at 65 °C for different times, respectively. As shown in Fig. 7b, the half-life curve of WT, D93C/F96C and K153C/S251C was y = −0.05913x + 3.4361 (R2 = 0.98), y = −0.04598x + 3.6209 (R2 = 0.99) and y = 0.04898x + 3.3817 (R2 = 0.97), respectively. Since the slope of the half-life curve is the inactivation rate constant kd, the t1/2 value of WT at 65 °C can be calculated as 11.7 min. The t1/2 of D93C/F96C and K153C/S251C was 15.1 and 14.2 min, respectively, which were 3.4 and 2.5 min higher than that of WT. The above results showed that the heat resistance of CPA was enhanced after the introduction of the disulfide bond, and the activity could be maintained for a longer period at a higher temperature.
The half-inactivation temperature (T50) refers to the temperature at which the activity drops to 50% of the initial activity. In this study, the recombinant WT, D93C/F96C and K153C/S251C was first subjected to different temperatures for a certain period, respectively. From the half-inactivation temperature curves (Fig. 7c), it can be seen that the activity of WT and its mutants decreased with increasing temperature, but the activity of WT decreased more significantly. After 15 min of treatment at 70 °C, the activity of WT was only 35% of the initial activity, whereas the mutants D93C/F96C and K153C/S251C were able to maintain about 50% of the initial activity. The T5015 of WT was calculated to be 58.0 °C, while the T5015 of mutants D93C/F96C and K153C/S251C were 66.5 and 69.4 °C, which were 8.5 and 11.4 °C higher than that of WT, respectively. The above results indicated that mutants D93C/F96C and K153C/S251C were more stable and could maintain better activity at higher temperatures.
Kinetic parameter measurements of recombinant CPA and its mutants
-
The enzyme kinetic constants, Km and Kcat/Km, were measured to evaluate the enzymatic properties of WT and its mutants. The Michaelis constant Km reflects the affinity of the enzyme for the substrate. The smaller the value is, the better the affinity of the enzyme for the substrate. As shown in Table 1, the Km of D93C/F96C and K153C/S251 were not significantly different from those of WT, indicating that the mutation did not affect the affinity for the substrate. The Kcat/Km denotes the catalytic efficiency of the enzyme. And, the larger the value is, the higher the catalytic efficiency of the enzyme is. As shown in Table 1, the Kcat/Km of D93C/F96C was the largest, whose catalytic efficiency was increased by 43.15% compared with that of WT. While, the Kcat/Km value of K153C/S251 was decreased by 8.08%, demonstrating that its catalytic efficiency was slightly lower than that of WT.
Table 1. Kinetic parameters of wild-type and mutant carboxypeptidase A.
Enzyme Km (μM) Vmax (μM·min−1) Kcat/Km (μM−1·s−1) WT 0.277 ± 0.012 1.833 ± 0.014 4.549 × 10−3 D93C/F96C 0.271 ± 0.021 2.569 ± 0.036 6.512 × 10−3 K153C/S251C 0.268 ± 0.043 1.641 ± 0.047 4.209 × 10−3 The above results of recombinant WT, D93C/F96C and K153C/S251C were similar to those of specific enzyme activities. It indicated that D93C/F96C achieved a dual improvement in thermostability and activity. Zhou et al. also found that the stability and activity of LPMOs could be improved simultaneously[11]. While the catalytic activity of K153C/S251C suffered from undesired effects when engineering thermostability, exhibiting a prevalent phenomenon 'stability-activity trade-off'[25]. Ming et al. also reported a similar result that the OTA degradation ability of CPA mutant R124K and S134P decreased to varying degrees while improving their thermostability[21].
Advanced structure evaluation of recombinant CPA and its mutants
-
The circular dichroic absorption spectra of recombinant CPA at 200~260 nm were scanned to investigate the effect of mutations on CPA secondary structure. Subsequently, the percentage content of secondary structures such as α-helixes, β-strands, β-turns and coils were determined by CDpro computer software. As shown in Fig. 8, the contents of α-helixes, β-strands, β-turns, and coils for WT were 24.6%, 26.6%, 21.6%, and 27.5%, respectively. Compared with WT, the mutants D93C/F96C and K153C/S251C showed an increase in α-helixes by 5.1% and 10.7%, while a decrease in β-strands, β-turns and coils. The above results indicated that the increase of α-helixes might be a key cause of their structural preservation[19], which was conducive to the improvement of the thermostability of CPA.

Figure 8.
The contents of secondary structures (α-helix, β-strand, β-turn, and coil) in CPA and its mutants.
The fluorescence emission spectra of recombinant CPA at 290~500 nm was scanned to investigate the effect of mutation on CPA tertiary structure. As shown in Fig. 9, WT and its mutants D93C/F96C and K153C/S251C all had a maximum emission at around 335 nm, demonstrating the characteristics of tryptophan fluorescence[28]. It suggested that there was no obvious impact on the tertiary structure of CPA when introducing disulfide bonds. A similar result was reported in 1,4-α-glucan branching enzyme. Introducing disulfide bonds might only effect the secondary structure of it while the tertiary structure continued a similar trend[29].
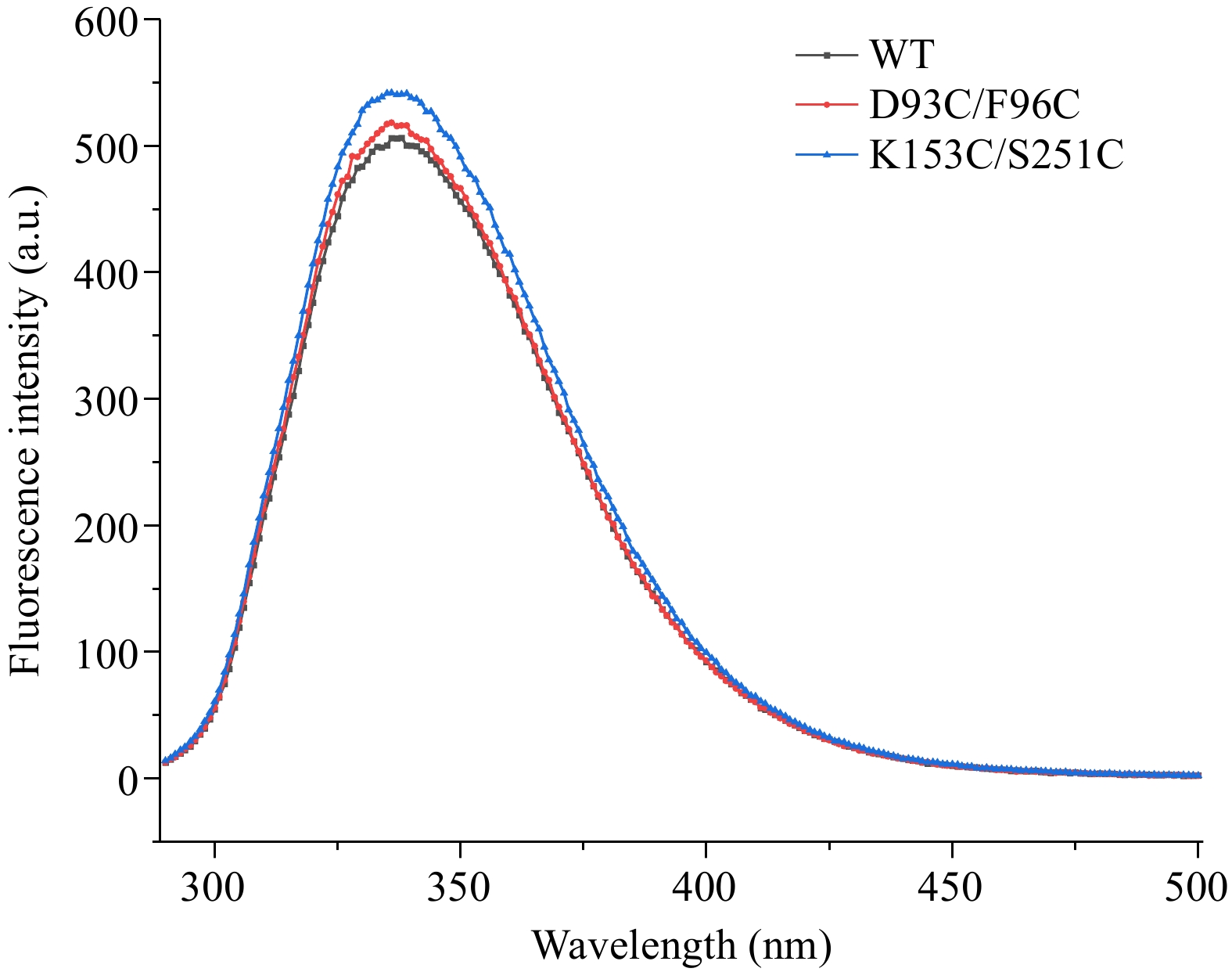
Figure 9.
Intrinsic fluorescence spectra of wild-type and mutant carboxypeptidase A.
Molecular interactions analysis of recombinant CPA and its mutants
-
The formation of disulfide bonds was quantified by detecting the amount of free sulfhydryl groups by DNTB[30]. As shown in Table 2, it was determined that WT possessed no natural disulfide bond. While, the mutants D93C/F96C and K153C/S251C both had one disulfide bond. The above results indicated that the mutants D93C/F96C and K153C/S251C successfully formed intramolecular disulfide bonds, realizing the purpose of enhancing the thermostability of CPA by introducing disulfide bonds.
Table 2. Comparison of disulfide bond number between wild-type and mutant carboxypeptidase A.
Enzyme A1 A2 Disulfide bond WT 0.456 ± 0.009 0.342 ± 0.010 0 D93C/F96C 1.434 ± 0.035 0.367 ± 0.018 1 K153C/S251C 1.549 ± 0.050 0.464 ± 0.023 1 The mutation of residues often leads to complex changes in multiple intramolecular interactions. In addition to analyzing disulfide bonds, ProteinTools online software was conducted to investigate the changes in the number of hydrogen bonds in mutant regions. Compared with WT, the mutant D93C/F96C showed an increase from one to two hydrogen bonds at mutation sites 93 and 96, increasing the thermostability by maintaining the rigidity of the enzyme structure (Fig. 10a). While, the mutant K153C/S251C lost the hydrogen bond formed between the mutation site 153 and 251, which might result in the decrease of enzyme activity (Fig. 10b). This was because hydrogen bonds were important factors for stabilizing the enzyme secondary structure[6].
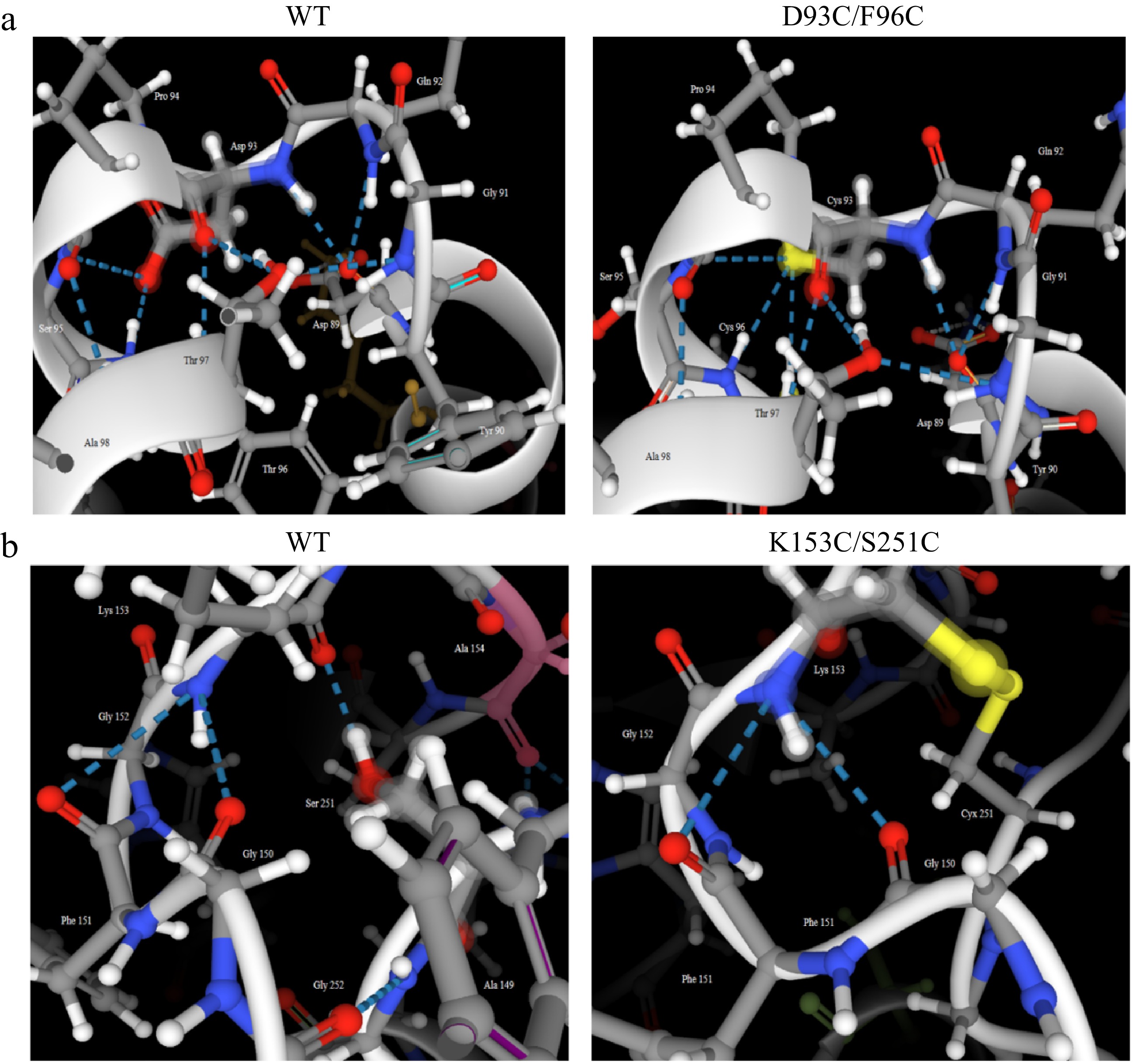
Figure 10.
Comparison of the number of hydrogen bonds in mutant regions between wild-type and its mutant carboxypeptidase A. (a) WT and D93C/F96C. (b) WT and K153C/S251C.
The changes in surface charges for WT and its mutants were analyzed by PyMOL software. The enhanced thermostability of D93C/F96C and K153C/S251C might also be related to the surface charge redistribution in mutant regions. As shown in Fig. 11a, the WT was electrically neutral at the sites of Asp93 and Phe96, with the region of Asp93-Phe96 showing electronegative. Whereas residues at sites 93 and 96 were both mutated to Cys, the mutant D93C/F96C exhibited positive electronegativity at sites 93 and 96. And, the mutant region shifted from negative to positive electronegativity. The phenomenon might be caused by the reduction of a carboxyl group and the formation of two sulfhydryl groups in residue structures. For mutant K153C/S251C, when replacing positively charged Lys at site 153 and positively charged Ser at site 251 with uncharged Cys, not only did the surface charges of mutation sites change from positive to neutral, but also the surface charges of the mutation region Lys153-Ser251 showed a similar trend (Fig. 11b). The disappearance of an amino group and a hydroxyl group and the formation of two sulfhydryl groups could be associated with the changes in surface charge of K153C/S251C. Chen et al. also reported a similar result that the redistribution of surface electrostatic charges enhanced the thermostability of glycosyltransferase UGT76G1[30]. Besides, Arabnejad et al. found that the positive effect on thermostability of halohydrin dehalogenase D162T could be contributed to the redistribution of surface electrostatic charges caused by the removal of the carboxyl group[31].
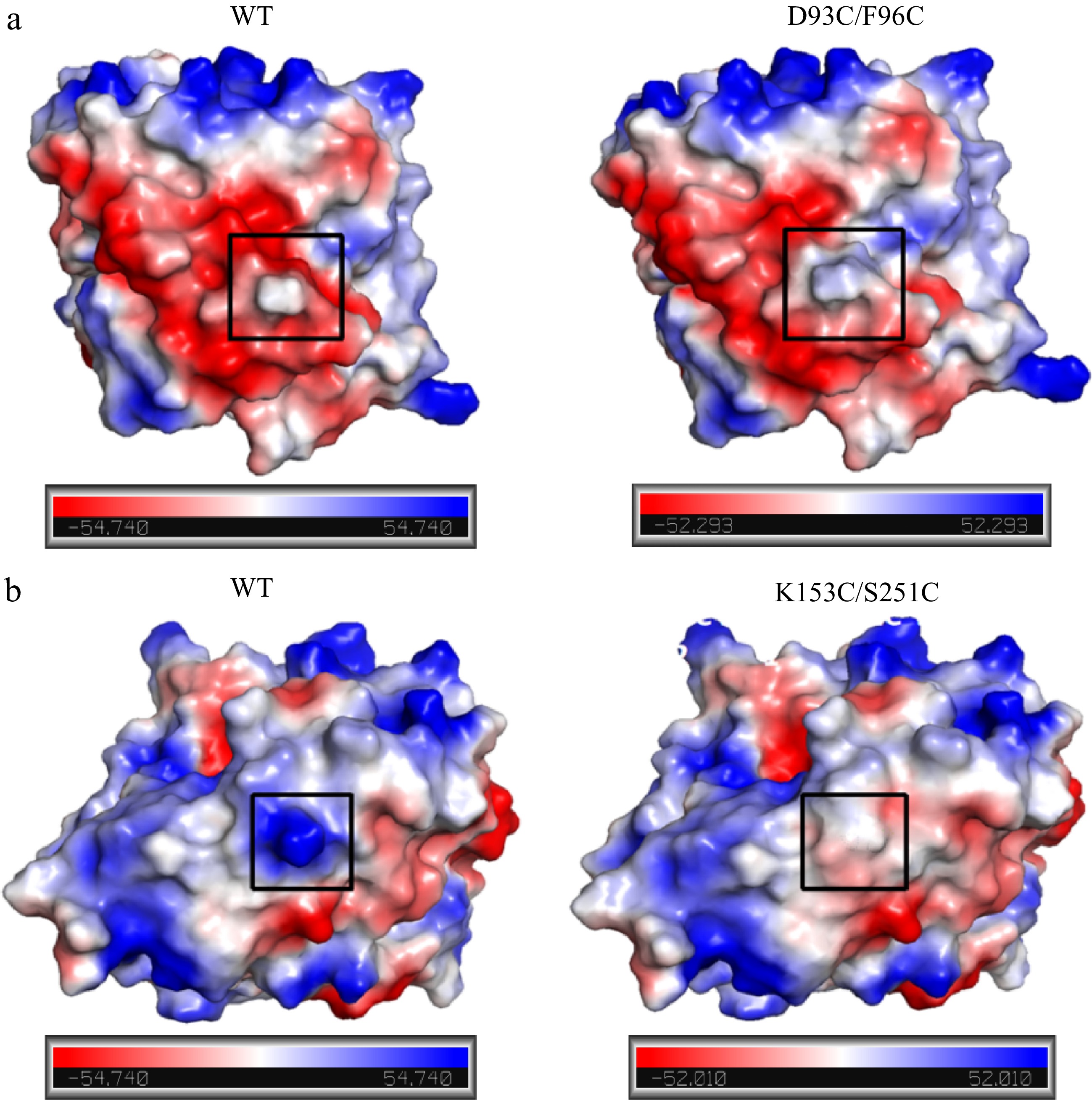
Figure 11.
Comparison of surface charge distribution in mutant regions between wild-type and mutant carboxypeptidase A. (a) WT and D93C/F96C; (b) WT and K153C/S251C. The mutation regions were circled in black wireframes.
-
A rational design of disulfide bonds was employed to enhance CPA thermostability. Two CPA mutants, D93C/F96C and K153C/S251C, were successfully designed and experimentally proved to possess enhanced thermostability. The mechanism of enhanced thermostability was related to changes in secondary structure and intramolecular interactions. This study showed that the rational design of disulfide bonds was an effective strategy to enhance the thermostability of CPA, which was helpful in broadening the applicability of CPA in industrial fields such as OTA degradation, bitter taste removal and so on.
-
The authors confirm contribution to the paper as follows: study conception and design: Zhang H, Liang Z; data collection: Zhang H, Zhao Z; analysis and interpretation of results: Zhang H, Zhao Z, Liang Z; draft manuscript preparation: Zhang H, Liang Z. All authors reviewed the results and approved the final version of the manuscript.
-
All data generated or analyzed during this study are included in this published article.
This research was funded by the National Key Research and Development Program of China (No. 2023YFD1301000); and the Shandong Province Natural Science Foundation (No. ZR202102260301).
-
The authors declare that they have no conflict of interest.
- Supplemental Fig. S1 The residue pairs forming potential disulfide bridges of carboxypeptidase A.
- Supplemental Fig. S2 The Sequence conservation of carboxypeptidase A.
- Supplemental Fig. S3 The ERRAT evaluation of mutant carboxypeptidase A D93C/F96C (a) and K153C/S251C (b).
- Supplemental Fig. S4 The Ramachandran plots of mutant carboxypeptidase A D93C/F96C (a) and K153C/S251C (b).
- Supplemental Fig. S5 Screening His+ transformations by MD plates of WT (a), D93C/F96C (b) and K153C/S251C (c).
- Supplemental Fig. S6 Screening of high-copy strains by G418 with gradient concentrations. (a) WT. (b)D93C/F96C. (c) K153C/S251C.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of China Agricultural University, Zhejiang University and Shenyang Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zhang H, Zhao Z, Zhu M, Logrieco AF, Wang H, et al. 2024. Enhancing the thermostability of carboxypeptidase A by rational design of disulfide bonds. Food Innovation and Advances 3(2): 191−201 doi: 10.48130/fia-0024-0017 

Enhancing the thermostability of carboxypeptidase A by rational design of disulfide bonds
- Received: 14 March 2024
- Revised: 04 June 2024
- Accepted: 05 June 2024
- Published online: 25 June 2024
Abstract: Carboxypeptidase A(CPA) has a great potential application in the food and pharmaceutical industry due to its capability to hydrolyze ochratoxin A(OTA) and remove the bitterness of peptide. However, CPA is a mesophilic enzyme that cannot adequately exert its catalytic activity at elevated temperatures, which seriously restricts its industrial application. In this study, the rational design of disulfide bonds was introduced to improve the thermostability of CPA. The highly flexible regions of CPA were predicted through the HotSpot Wizard program and molecular dynamics (MD) simulations. Then, DbD and MODIP online servers were conducted to predict potential residue pairs for introducing disulfide bonds in CPA. After the conservativeness analysis of the PSSM matrix and the structural analysis of the MD simulation, two mutants with potentially enhanced thermostability were screened. Results showed that these mutants D93C/F96C and K153C/S251C compared to the wild-type(WT) exhibited increase by 10 and 10 °C in Topt, 3.4 and 2.7 min in t1/2 at 65 °C, in addition to rise of 8.5 and 11.4 °C in T5015, respectively. Furthermore, the molecular mechanism responsible for thermostability was investigated from the perspective of advanced structure and molecular interactions. The enhanced thermostability of both mutants was not only associated with the more stable secondary structure and the introduction of disulfide bonds but also related to the changes in hydrogen bonds and the redistribution of surface charges in mutant regions. This study showed for the first time that the rational design of disulfide bonds is an effective strategy to enhance the thermostability of CPA, providing in this way a broader industrial application.
-
Key words:
- Disulfide bonds /
- Thermostability /
- Carboxypeptidase A /
- Rational design