








-
As societal standards have risen, hypertension has emerged as a significant chronic health concern, affecting approximately 30% of the adult population in most countries[1,2]. Furthermore, it has been identified as the leading cause of mortality worldwide, accounting for 12.8% of all deaths[2]. The population of hypertension patients is expected to increase to 1.5 billion by 2025[3]. Despite synthetic drugs being a standard treatment for hypertension, 50% of patients still have blood pressure that is not adequately controlled[4]. In addition, the long-term use of drugs will produce potential side effects, increase patients' non-compliance to drugs, and endanger patients' health[5]. This has led to a new trend to find food-derived active peptides that effectively inhibit hypertension instead of chemical synthetic agents[6].
Angiotensin-converting enzyme (ACE) was isolated in 1956 and used as a 'hypertension-converting enzyme'[7]. ACE is a chloride and zinc-dependent dipeptidase that plays a vital role in the regulation of electrolyte balance, blood pressure regulation, cardiovascular system development, and vascular remodeling by converting angiotensin I (Ang I) to angiotensin II (Ang II) in body fluids[8]. The potential of ACE inhibitory peptides to lower blood pressure has been the subject of considerable research. The peptides have been found to exert this effect by inhibiting the binding between Ang II and the receptor, or by increasing nitric oxide (NO) levels[6,9]. For instance, in cellular experiments conducted by Qiao et al., it was demonstrated that the ACE inhibitory peptides derived from tuna effectively suppressed the binding of Ang II to its receptor on ACE through an increase in NO content[9]. Wei et al. identified two ACE inhibitory peptides, FDRPFL and KWEKPF, by enzymatic digestion of Chinese Rushan cheese by-product[10]; Lin et al. investigated the digestion products of earthworm proteins, from which they isolated seven ACE inhibitory peptides SSPLWER, RFFGP, LERWP, MFPGIADR, FPGIADR, SADRISHGF, and ADRYSSWP[11]; Hu et al. isolated three ACE inhibitory peptides, SEGPK, FDGPY, and SPGPW, from Monkfish (Lophius litulon) swim bladders[12]; Suo et al. employed alkaline protease for the preparation of tuna byproducts-milts hydrolysates and identified ten ACE inhibitory peptides[13].
Seabuckthorn is a premium source of bioactive peptides, given its comprehensive range of amino acids and elevated proportion of essential amino acids[14]. The total protein content of various seabuckthorn from India has been reported to range from 46 to 129 g/kg dry weight (DW). The protein content in the Polish seabuckthorn variety is 93 g/kg DW[15]. It is characterized by high protein content compared to other berries such as strawberry (Fragaria ananassa Duch.) (9 g/kg DW), golden berry (Solanum mammosum L.) (18.5 g/kg DW)[16], raspberry (Rubus idaeus L.) (33−44 g/kg DW)[17]. Seabuckthorn seed meal is a by-product generated during the supercritical CO2 extraction of seabuckthorn oil. Currently, most of this by-product is employed as animal feed or discarded as waste, resulting in a significant loss of resources. The objective of this study is to extract proteins and peptides from seabuckthorn seed meals that inhibit ACE activity, thereby increasing the utilization of seabuckthorn seed meal, reducing the waste of resources, and providing an alternative option for hypertensive patients.
In this paper, seabuckthorn seed meal protein was hydrolyzed, investigated its inhibitory activity for ACE, determined its inhibitor value, and characterized its structure. Finally, ten peptides with ACE inhibitory activity were identified by mass spectrometry and further elucidated the mechanism of its interaction at the molecular level through molecular docking.
-
The seabuckthorn seed meal was provided by Puredia Limited (Xining, Qinghai, China) and was identified to contain 30.80 g of protein per 100 g of seed meal; Sunflower oil purchased from Yihai Kerry Arawana Holdings Co. (Shanghai, China); Compound enzyme preparation ZF101 from Angel Yeast Co. (Yichang, Hubei, China); Angiotensin-converting enzyme (ACE), and Hippuryl-histidyl-leucine (HHL) from Sigma-Aldrich Chemical Co. (Beijing, China); Protein Content Assay Kit, Amino Acid (20AA), Pepsin (1:3000), Trypsin (1:250) from Beijing Solarbio Science & Technology Co. (Beijing, China), Sodium hydroxide, hydrochloric acid from Tianjin Damao Chemical Reagent Factory (Tianjin, China).
Preparation of protein from seabuckthorn seed meal
-
The seabuckthorn seed meal was crushed and screened using a 120-mesh sieve, seabuckthorn seed meal with a mesh size greater than 120 continued to be crushed until all samples passed through the sieve. The screened seabuckthorn seed meal was added with water to make a suspension with a material-to-liquid ratio of 1:30. The pH of the system was adjusted to 10 with 1 M NaOH, and the pH of the system was maintained at this level. The extraction was conducted using a magnetic stirring water bath (SHJ-2A, Changzhou Jintan Liangyou Instrument Co., Limited) at 60 °C for 80 min. Once the extraction process was complete, the supernatant was collected and the pH was adjusted to 3 with 1 M HCl. The solution was then allowed to stand for 30 min, after which centrifugation was performed at 1,043 g for 10 min. The precipitate, which constituted the seabuckthorn seed meal protein, was then lyophilized for subsequent use.
The formula for protein yield calculation:
$ Protein\;yield\;\left(\text{%}\right)=\dfrac{{m}_{1}}{m}\times 100 $ where, m1: the amount of protein extracted; m: the quality of seabuckthorn seed meal used.
Preparation of ACE inhibitory peptides
-
The protein of seabuckthorn seed meal was dissolved in water and prepared into a 7% (w/v) protein solution. Enzymolysis with 4.2% (w/w) compound enzyme preparation ZF101 was performed at 55 °C for 6.38 h. According to Li et al.[18], it has been demonstrated that ACE inhibitory activity is more pronounced for peptides smaller than 3 kDa. The enzyme solution was filtered through a 5 μm filter bag and then a 3 kDa filter membrane. The enzyme hydrolysate less than 3 kDa was retained for analysis.
Determination of molecular weight (MW) distribution of proteins and peptides
-
Sample pretreatment
Approximately 100 mg of the sample was placed in a 10 mL volumetric flask, then the mobile phase was sucked into the volumetric flask until it reached the calibration line, then processed with 100 W/L ultrasonic density for 5 min, centrifuged at 261 g, filtered with microporous filter membrane, and waited for sample injection.
High-performance gel chromatography was employed to ascertain the molecular weight distribution of peptides and seabuckthorn seed meal proteins. The Waters 2695 HPLC (equipped with a 2487 UV detector and Empower workstation GPC software) was employed to ascertain the chromatographic conditions. The TSKgel 2000 SWXL column (Tosoh Biotechnology Co., LTD, Shanghai, China) was 300 mm × 7.8 mm in diameter and the mobile phase consisted of acetonitrile/water/trifluoroacetic acid in a ratio of 45/55/0.1 (V/V). Detection was carried out using a wavelength of 220 nm, with a flow rate of 0.5 mL/min, and the temperature of the column was set at 30 °C (Sigma Aldrich Trading Co., Ltd., Shanghai, China). The following compounds were used as standards for the molecular weight correction curve of seabuckthorn seed meal proteins (Sigma Aldrich Trading Co., Ltd., Shanghai, China): cytochrome C (MW 12384), trypsin inhibitor (MW 6500), bacitracin (MW 1422), Gly-Gly-Tyr-Arg (MW 451), and Gly-Gly-Gly (MW 189). Bovine albumin (MW 67000), ovalbumin (MW 45000), chymotrypsinogen A (MW 24000), and lysozyme (MW 14300).
Determination of peptide yield
-
The trichloroacetic acid nitrogen solubility index method (TCA-SNI) was employed, with slight modification. Insoluble proteins in solution were precipitated with trichloroacetic acid, and following centrifugation, the nitrogen content in the supernatant was determined using the Kjeldahl nitrogen method[19]. The specific method was to take 1 mL of protein hydrolysate, add 3 mL trichloroacetic acid solution, shake and mix for 15 min, then centrifuge at 1,043 g for 10 min, take the supernatant, and use a Protein Content Assay Kit (Biuret Method) to determine peptide content.
The formula for the calculation of polypeptide yield:
$ Yield\;of\;peptides\;\left(\text{%}\right)=\dfrac{{n}_{1}}{n}\times 100 $ where, n1: peptide content in the supernatant; n: the amount of protein consumed.
Analysis of peptide structure
Amino acid composition analysis
-
With certain modifications based on the methodology proposed by Zhu et al.[20]. A quantity of the sample was subjected to hydrolysis with 6 M HCl at 110 °C for 22 h, then removed and neutralized with NaOH. Following this, it was filtered through a 22 μm membrane and assayed. HPLC parameters (Waters2695, equipped with 2487 UV detector and Empower workstation GPC software, Agilent, USA), chromatographic conditions: Column: C18 SHISEIDO (Shanghai Inguang Trading Co., Ltd., Shanghai, China) (4.6 mm × 250 mm × 5 μm); injection volume: 10 μL; column temperature: 40 °C; The flow rate was stabilized at 1.0 mL/min. UV detection conditions were as follows: the wavelength was 338 nm and changed to 262 nm at 22.5 min. Fluorescence detector conditions: excitation wavelength 340 nm, emission wavelength 450 nm, 22.5 min excitation wavelength changed to 266 nm, emission wavelength 305 nm; Mobile phase: A: sodium acetate-triethylamine-tetrahydrofuran solution, B: sodium acetate-acetonitrile-methanol solution; Standard: 17 amino acids (Sigma Aldrich Trading Co., ltd., Shanghai, China).
Infrared spectral analysis
-
The sample lyophilized powder (2 mg) was mixed with 200 mg of dried KBr. The mixture was then ground evenly with an agate mortar, pressed, and scanned with an infrared spectrometer (VERTEX70, VERTEX, Germany) with a KBr background in the spectral range 4,000−500 cm−1. Software Origin 2019 was used for mapping, and PeakFit v4.12 software was used for analysis. Two-point baseline correction was performed in the spectral range 1,600−1,700 cm−1, followed by Savitsky-Golay function smoothing, and fitted in second derivative spectra. Manual fitting can be performed multiple times to reduce the residual error (R2 ≥ 0.999). The percentage of secondary structure based on the area of each peak was calculated[21].
X-ray diffraction analysis
-
Adjustments were made according to Lin et al.[22]. Samples were examined using an X-ray diffractometer (D8ADVANCEA25, Bruker, Germany) at a voltage of 40 kV and a current of 40 mA with a step scan of 0.02° from 5° to 70°.
Analysis of peptides and properties
-
Peptides were identified by Quadrupole-TOF LC-MS/MS System. The analytical column used in capillary liquid chromatography: Mobile phase A: 0.1% formic acid; Mobile phase B: 0.1% formic acid and 80% acetonitrile; Flow rate: 300 nL/min. Primary mass spectrometry parameters: Resolution: 60000; AGC target: 4.0e5; Maximum IT: 100 ms; Scan range: 375−1,500. Secondary mass spectrometry parameters: Resolution: 15,000; AGC target: 5.0e4; Maximum IT: 256 ms. The range scan mode is m/z Normal (Auto), and the HCD Collision Energy (%) is 30. The isoelectric point (pI), net charge, and solubility of peptides were calculated using the method used by Lear & Cobb[23].
Determination of IC50
-
The borate buffer was prepared with a pH of 8.3, an 8 mM HHL solution, and an ACE solution with an activity of 0.1 U/mL. Three test tubes were prepared and designated as sample A, control B, and blank C. Each test tube was then supplied with 200 mL of the HHL solution, 250 mL of HCl to test tube C and 100 mL of the enzymatic hydrolysate. Test tube A was placed in a water bath set at 37 °C for 5 min. Subsequently, 20 mL of ACE solution was added to each of the three test tubes, which were then incubated in the water bath for an additional 30 min. Then, an HCl solution was added to test tubes A and B, and a borate buffer solution was added to test tubes B and C to make them react. After the reaction, 1.7 mL ethyl acetate was added to each tube and full reaction 20 s before allowing it to stand until the liquid stratified. 1 mL of the ethyl acetate layer was removed from each tube and placed into a drying oven at 120 °C for 30 min. After drying, the tube was removed and the solution was prepared in 3 mL of ultra-pure water. The UV spectrophotometer was then calibrated to 228 nm to measure the absorbance value of the dissolved solution[24−26]. The enzymatic solution was diluted to five concentration gradients, and the measured results were analyzed by IC50. The calculation formula is as follows:
${\rm ACE\; inhibition\; rate\; ({\text{%}})} =\dfrac{{A}_{B}-{A}_{A}}{{A}_{B}-{A}_{C}}\times 100 $ where, AB: Absorbance of the control tube; AA: Absorbance of the sample tube; AC: Absorbance values of blank tubes.
Kinetics of ACE inhibition
-
Peptides less than 3 kDa were diluted and divided into three concentrations: 19.5, 9.75, and 0 mg/mL. The reaction was carried out at four different concentrations of HHL (8.0, 4.0, 2.0, and 0.8 mmol/L, respectively). The reciprocal of the reaction rate (1/V) is used as the ordinate and the reciprocal of the substrate concentration (1/[S]) as the abscissa Lineweaver-Burk double reciprocal plot was made to analyze the inhibition pattern, and the inhibition constants Km and Vmax were calculated by the intercept between the line and the horizontal and vertical coordinates[27,28].
Stability of ACE inhibition
-
According to Wang's team[29], the stability of peptides at different temperatures and pH was determined. Peptide solutions with a molecular weight of less than 3 kDa were subjected to a water bath treatment at temperatures of 4, 20, 40, 80, and 100 °C for 2 h. Following this, the inhibitory rate on ACE was determined. The peptide solution of less than 3 kDa was kept at pH 2, 4, 6, 8, 10, and 12 at 37 °C for 2 h, and the pH was adjusted to 8.3 to determine the ACE inhibition rate of the peptide.
Molecular docking
-
The 1O8A crystal in the RCSB PDB database (PDB DOI: 10.2210/pdb1O8A/pdb) was used as the acceptor ACE structure in this section, and the Zn and Cl atoms in 1O8A were retained and saved in pdb format for use during docking. The molecular structure of the ACE inhibitory peptide was plotted using the Chemdraw18.0 software, and the energy of the molecular structure was minimized. The molecular docking was performed with the software AutoDockTools-1.5.6, and the center coordinates of the docking box were 40.575 in x, 37.430 in y, and 43.759 in z. The Analyze program in AutoDockTools-1.5.6 software was used to view and analyze the results, and PyMOL software was used to plot the docking results for ACE and peptides.
Statistical analysis
-
All samples were measured at least three times, and data were represented as average ± standard deviation. The data were analyzed using IBM SPSS Statistics 27.
-
The preparation of seabuckthorn seed meal peptides is shown in Fig. 1. Seabuckthorn seed meal, which is a byproduct of the oil extracted from seabuckthorn seeds, contains a considerable concentration of protein and a plethora of amino acids. This quality renders seabuckthorn seed meal a superior source of protein[30]. In this experiment, seabuckthorn seed meal was crushed, and seabuckthorn protein was extracted by alkaline extraction and acid precipitation, and the protein yield was 27.66%. The protein extraction rate increased with the increase in pH. However, too high a pH leads to the production of a toxic lysinoalalnine protein that leads to the passivation of metalloenzymes and nephrotoxicity and further compromises the nutritional value of foods[31]. To better preserve the flavor, color, structure, and nutrition of seabuckthorn seed meal protein, freeze-drying was selected[32,33]. To optimize the industrial production of seabuckthorn seed meal peptide, a food-grade enzyme preparation was used. The optimal pH for enzyme activity was neutral, which reduced the production of inorganic salts and avoided the loss of equipment in the production process. After enzymatic digestion with ZF101 protein, the yield of polypeptides obtained from seabuckthorn seed meal was 24.29%.
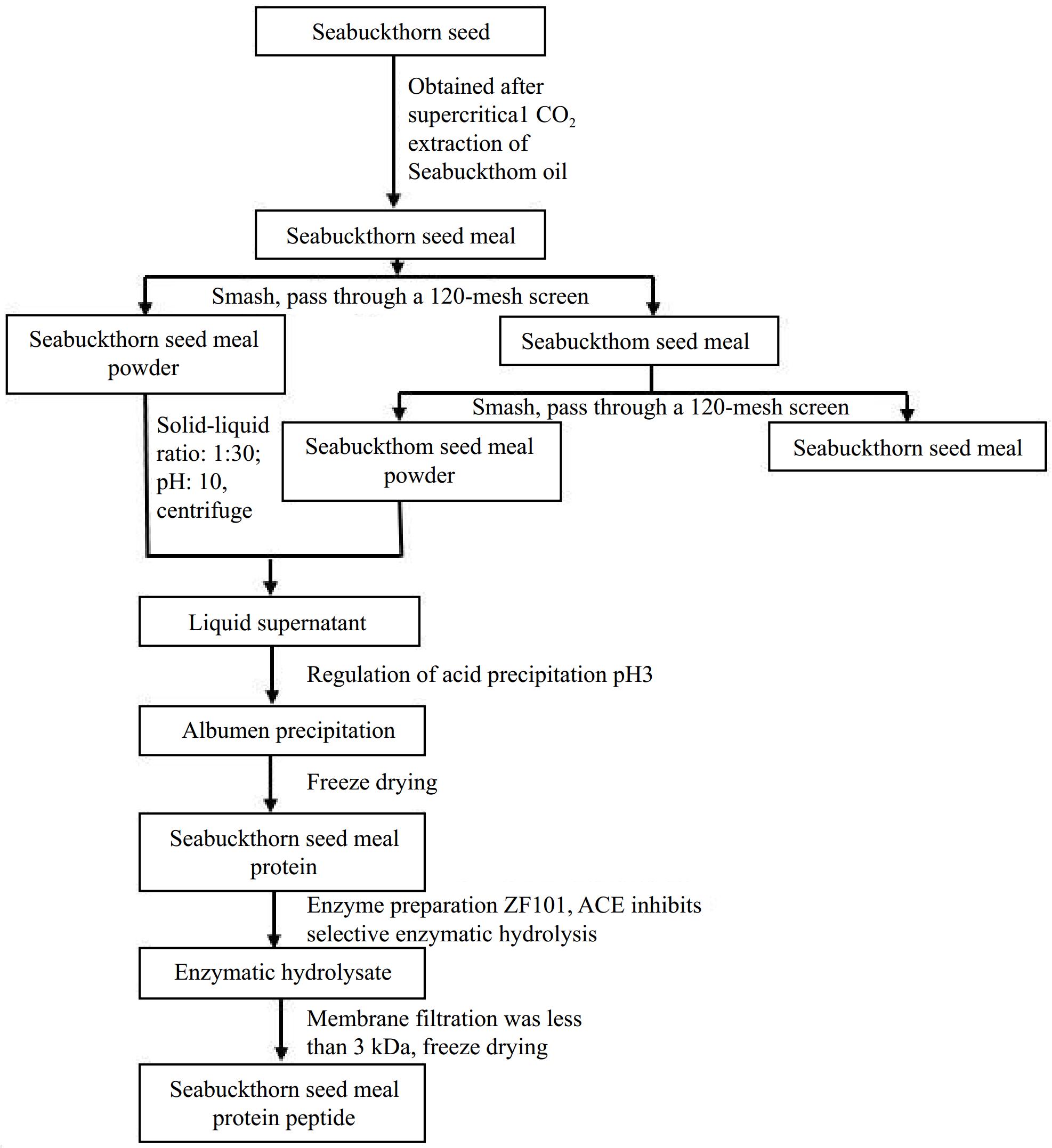
Figure 1.
Preparation of ACE inhibitory peptides from seabuckthorn seed meal.
MW distribution of proteins and peptides
-
The MW distribution of seabuckthorn seed meal protein before and after enzymatic digestion is shown in Tables 1 & 2. Table 1 shows that the molecular weight of seabuckthorn seed meal protein is relatively large, mainly distributed in the range of 50~10 kDa, with an average molecular weight of 170,280 Da. Following enzymatic hydrolysis, the molecular weight of peptides is markedly diminished, with the average molecular weight reduced to 2,842 Da. Most of these peptides have a molecular weight below 3 kDa, representing 63.41% of the total. The free amino group (< 180 Da) was 14.44%. The content of peptides in the range of 180−3,000 Da was 48.97%.
Table 1. Test results of protein MW of seabuckthorn seed meal.
MW (Da) PAP (%) NAMW (Da) WAMW (Da) > 500 k 13.98 768,704 803,432 500 k~100 k 15.45 191,719 239,242 100 k~50 k 11.52 67,218 69,982 50 k~20 k 26.88 30,453 32,557 20 k~10 k 24.98 14,362 14,858 < 10 k 7.19 4,191 6,573 Table 2. Determination of polypeptide MW of seabuckthorn seed meal.
MW (Da) PAP (%) NAMW (Da) WAMW (Da) > 10000 3.26 11,507 11,678 10,000~5,000 19.40 6,760 7,012 5,000~3,000 13.93 3,871 3,956 3,000~2,000 8.84 2,448 2,482 2,000~1,000 12.38 1,401 1,458 1,000~500 11.09 698 725 500~180 16.66 283 304 < 180 14.44 / / MW, Molecular weight; PAP, peak area percentage; NAMW, number average molecular weight; WAMW, weight average molecular weight. Structure of polypeptides from seabuckthorn seed meal
Amino acid composition
-
Based on the analysis shown in Table 3, the polypeptide of seabuckthorn seed meal contains 17 amino acids, a total of 53.61 g/100 g, of which the proportion of essential amino acids is 27.88. According to the nature of amino acids, the content of acidic amino acids was the highest (39.33%), followed by hydrophobic amino acids (29.24%) and aromatic amino acids (5.88%). According to Cheung et al. and Suetsuna's & Nakano, the composition and content of amino acids play an essential role in ACE inhibition[34,35]. Hayes et al. found that hydrophobic amino acids, such as Phe, Trp, Tyr, Val, Leu, and Ile, favored to combine with the C-terminal active site of ACE[36]. Additionally, C-terminal amino acid residues containing positive charge, such as Lys and Arg, could be conducive to increasing the ACEi activity of peptides. Pro arouses great attention of researchers because Pro occurs frequently at the C-terminus of antihypertensive peptides, such as LYPPP, YSMYPP, VGLYP, EVSQGRP, YP, and KDEDTEEVP[1]. This is consistent with the amino acid composition analyzed in this assay.
Table 3. Amino acid composition analysis.
Name Time
(min)Area
(mau*s)Content
(g/100 g)Proportion of
total amino
acid contentAspartic acid (Asp) 2.953 696.862 6.366 11.873% Glutamic (Glu) 3.400 1327.706 14.757 27.525% Serine (Ser) 6.851 363.541 2.638 4.921% Histidine (His) 7.780 62.961 1.148 2.141% Glycine (Gly) 8.724 412.175 2.162 4.033% Threonine (Thr) 9.093 180.797 1.534 2.860% Arginine (Arg) 10.430 696.119 8.622 16.082% Alanine (Ala) 11.566 339.348 2.072 3.864% Tyrosine (Tyr) 13.934 90.650 1.239 2.311% Valine (Val) 17.986 294.825 2.415 4.505% Methionine (Met) 18.348 17.390 0.177 0.330% Phenylalanine (Phe) 20.963 162.056 1.918 3.577% Isoleucine (Ile) 21.412 217.560 1.984 3.701% Leucine (Leu) 22.678 399.479 3.569 6.657% Lysine (Lys) 23.488 243.452 1.468 2.739% Proline (Pro) 29.987 291.578 1.391 2.594% Cystine (Cys) 16.960 10.302 0.154 0.287% Infrared (IR) spectra of polypeptides
-
There is an essential relationship between the secondary structure of peptides and their intramolecular bonding. Fourier transform infrared spectroscopy is a powerful method. Figure 2a shows that the peptide contains C-H, N-H, C=O, C-N, and -COO functional groups according to the fluctuations in the IR spectra[37]. According to Zhao et al.[21], the band ranges of β-folding, random coil, α-helix, and β-angle were 1,600−1,640, 1,640−1,650, 1,650−1,660, and 1,660−1,700 cm−1, respectively, which were processed by software PeakFit v4.12. The results of the amide I band (1,600−1,700 cm−1) showed that the β-fold structure was the main secondary structure with a content of 61.87% ± 1.42%. The content of the β-turn structure was 38.13% ± 1.42%, while no other structural motifs were detected.
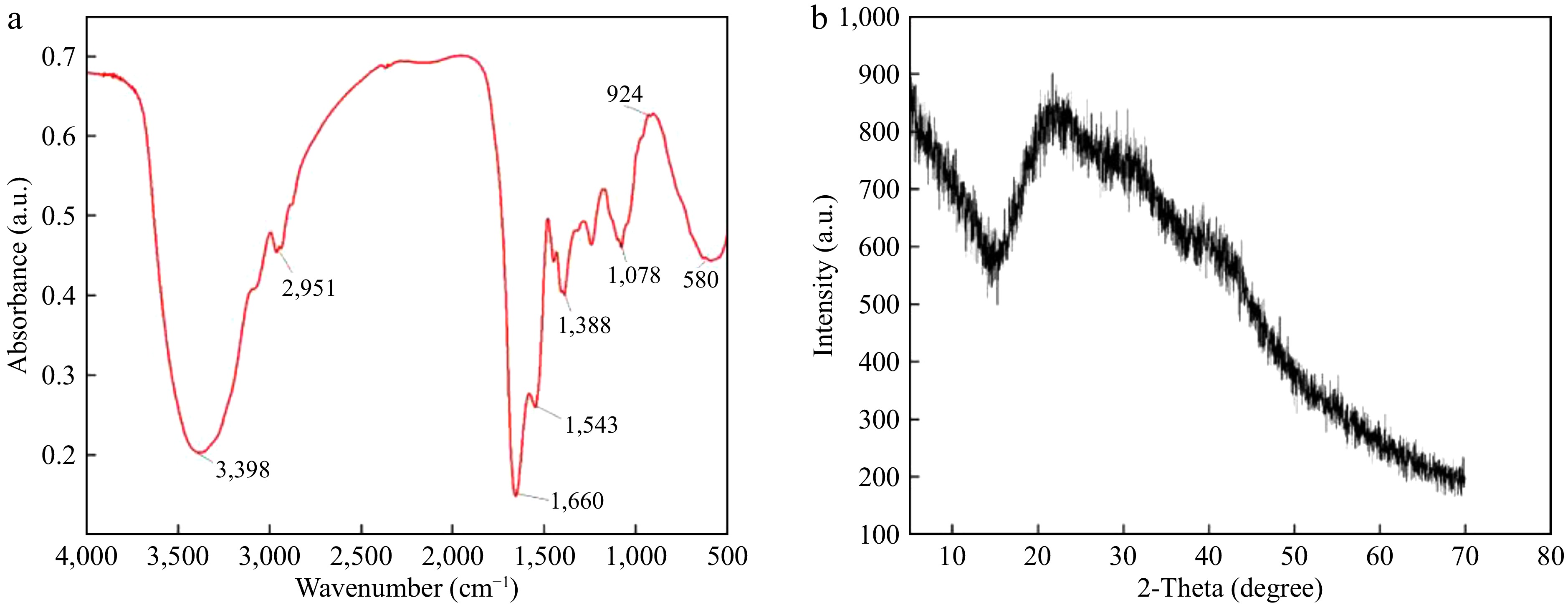
Figure 2.
(a) Infrared diffraction spectra of peptides. (b) X-ray diffraction results of peptides
X-ray diffraction of polypeptides
-
As can be seen in Fig. 2b, the sample has a diffraction angle of 22. The figure shows a diffraction absorption characteristic peak. However, the peak intensity is low, and the background is broad, which indicates that the ACE inhibitory peptide of seabuckthorn seed meal has a random amorphous structure.
Peptide identification and characterization
-
The detection results of peptides < 3 kDa were compared with the NCBI and Uniprot databases of ACE inhibitory peptides, and 10 ACE inhibitory peptides were detected. The results are shown in Table 4. The molecular distributions of the peptides measured in this study range from 1,000 to 3,000 Da. Except for the peptides AGGGGGGGGGGSRRL and DDEARINQLFL, which are slightly larger than 1 kDa, the rest of the peptides are more significant than 1.5 kDa, presumably because most of the peptides obtained by enzymatic hydrolysis are not recorded in the database and are not detected. Ma et al. and Toopcham's team arginine could enhance the ACE-inhibiting activity of peptides, and most of the peptides screened in this study contained arginine, similar to the amino acid sequence screened[38,39].
Table 4. Peptide detection results and physicochemical properties.
Serial number Amino acid sequence Mass-to-charge ratio Isoelectric point Static charge Solubility 1 FRVAWTEKNDGQRAPLANN 2187.105 pH 9.86 1 Good 2 LIISVAYARVAKKLWLCNMIGDVTTEQY 3198.705 pH 8.70 0.9 Poor 3 VIRSRASDGCLEVKEFEDIPP 2360.191 pH 4.16 −2.1 Good 4 AGGGGGGGGGGSRRL 1172.588 pH 12.10 2 Good 5 LQPREGPAGGTTALREELSLGPEAALDTPPAGP 3268.681 pH 3.88 −3 Good 6 DDEARINQLFL 1333.675 pH 3.54 −2 Good 7 FAVSTLTSYDWSDRDDATQGRKL 2632.264 pH 4.19 −1 Good 8 RQLSLEGSGLGVEDLKDN 1929.988 pH 3.93 −2 Good 9 GGGGGGGGGGGGGGGIGGGGGGGGGGGAR 1841.798 pH 10.84 1 Good 10 KEALGEGCFGNRIDRIGDVSGMGCNRRTPAP 3276.578 pH 8.07 0.9 Good Determination of IC50
-
The relationship between the concentration of ACE inhibitory peptide and the inhibition rate of seabuckthorn seed meal is shown in Fig. 3a (R2 = 0.9538). ACE inhibitory peptide concentration and inhibitory activity show a positive correlation. Based on the correlation curves in Fig. 3b, the IC50 is 4.358 mg/mL.
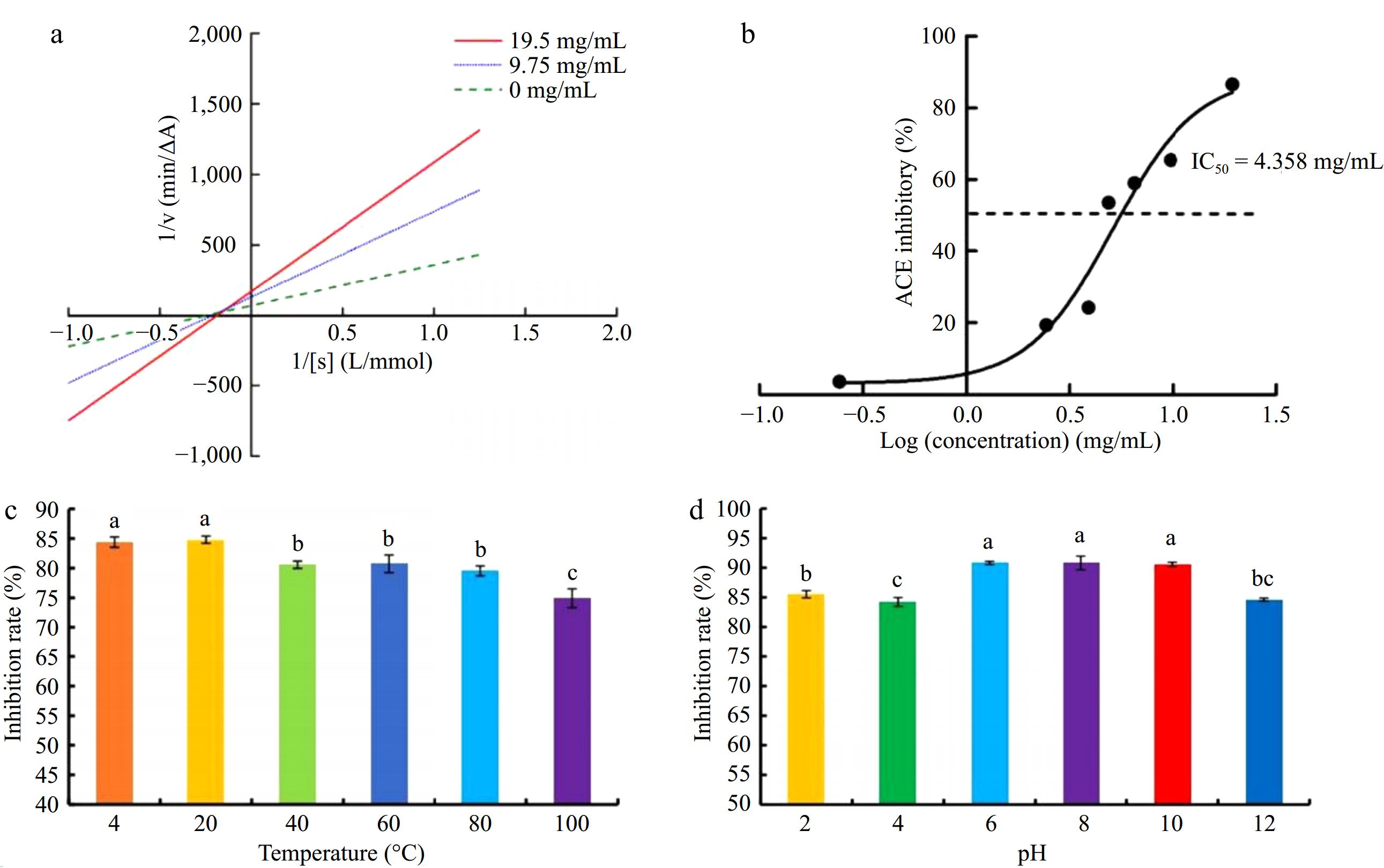
Figure 3.
(a) Relationship between ACE inhibitory peptide concentration and inhibitory activity of seabuckthorn seed meal. (b) Kinetic double reciprocal diagram of seabuckthorn seed meal polypeptide inhibiting ACE. (c) The ACE inhibition rate of seabuckthorn seed meal peptides treated at different temperatures. (d) The ACE inhibition rate of seabuckthorn seed meal peptides treated at different pH. Different letters represent significant differences at p < 0.05.
ACE inhibition mechanism of peptides
-
Lineweaver-Burk plots were used to analyze the kinetic mechanism of ACE inhibition of seabuckthorn seed meal peptides. As shown in Fig. 3a, the Lineweaver-Burk line of seabuckthorn seed meal peptide intersects 1/[S], indicating that the inhibition type of ACE by seabuckthorn seed meal peptide is non-competitive. This proved that the seabuckthorn seed meal polypeptide does not compete with the substrate and can bind to the ACE-HHL complex or sites other than the ACE active site, changing the spatial formulation of ACE and reducing the activity[40]. The ACE inhibition rate of the polypeptides from seabuckthorn seed meal was positively correlated with the concentration, similar to the inhibition mode of the ACE inhibitory peptide derived from tilapia skin gelatin[41]. The tetrapeptide FRVM from the algae[18], and all of them were non-competitive.
Inhibitory stability analysis of peptides
-
Figure 3c & d shows the results of the inhibition rate determined after treatment of peptide solution < 3 kDa at different temperatures and pH for 2 h. There was no significant change in the inhibition rate of ACE by the polypeptides of seabuckthorn seed meal, which indicated that peptides < 3 kDa had good thermal stability and pH stability.
The ACE inhibition rate of 1% (w/v) peptide solution was 67.46% ± 3.83%. The undigested peptide solution was digested by pepsin and trypsin, and its structure was destroyed. In general, the ACE inhibitory peptide of seabuckthorn seed meal peptides still has an excellent inhibitory ability after digestion. This suggests that the performance of the peptides is stable after oral administration[29]. Studies have shown that some ACE-inhibitory peptides do not achieve the desired effect after they enter the body because they are broken down into peptides with low ACE-inhibitory activity in the stomach[42,43].
Molecular docking simulation between peptides and ACE
-
Molecular simulation docking was performed on ten selected peptides with ACE inhibitory activity, and the docking results are shown in Fig. 4. According to Natesh's team[44], the active sites in ACE can be divided into three pockets, namely S1, S2, and S1′. S1 contains Glu384 and Tyr523. S2 contains His353, Ala354, Cys511, His513 and Tyr520. S1′ contains Gln162, Asp377, and a triplex of catalytic sites for His383, His387, Glu411, and Zn2+.
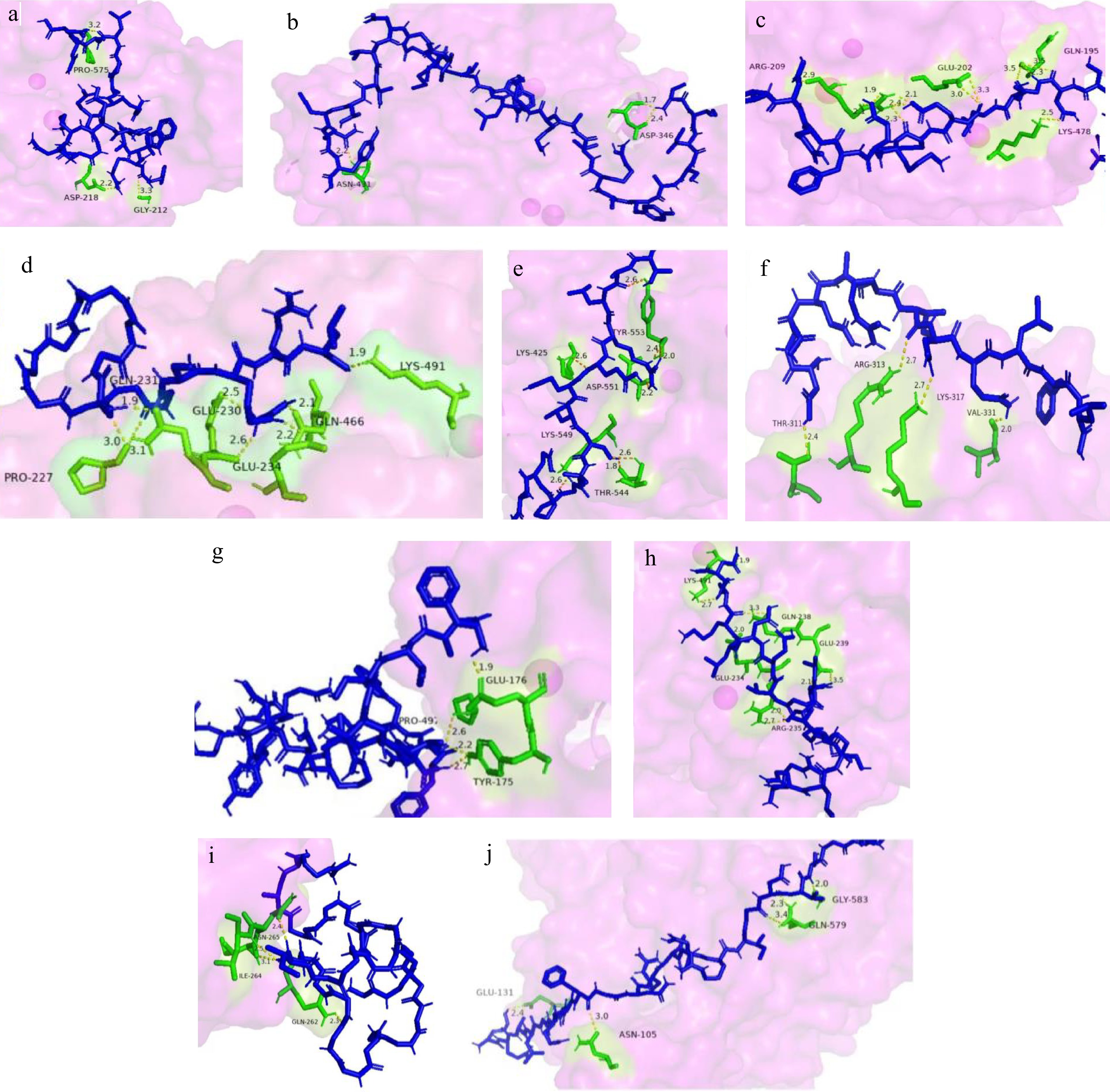
Figure 4.
Molecular docking results of peptides and ACE (1−10 docking results of 10 peptides in Table 2). Blue is peptide; Purple is ACE; Green is amino acid; Yellow is the bond created by the interaction.
Using simulated docking, it was found that ten peptides interact with ACE at different sites and form different numbers of hydrogen bonds with ACE. Only the fifth peptide and ACE interact in chain-to-chain, and no bonds are formed at the atomic level. The binding ability of inhibitory peptides and ACE is related to the number and length of hydrogen bonds they form. In the results of this simulation, peptide 3 forms many hydrogen bonds with ACE, including Arg209, Glu202, Gln195, and Lys478, to form 12 hydrogen bonds, six of which were formed with Arg209, followed by peptides 4 and 8. The binding sites of peptide 4 are Gln231, Pro227, Glu230, Glu234, Gln466, and Lys491. Although the binding sites are not located at the active center of the ACE, they are all located near Zn2+. Peptides 1 and 2 form the fewest hydrogen bonds (only three), and none of them are in the ACE active site, but they are still close to the active pocket. In addition to hydrogen bonding, interactions between peptide chains are also crucial in maintaining the stability of the complexes they form.
-
Over the past decade, there has been a growing interest in the extraction of bioactive peptides from food. There is a trend towards the screening of bioactive peptides for use as drug substitutes in the treatment of hypertension[6]. A significant number of papers have investigated the structural characterization, bioavailability, functional effects, and inhibitory mechanisms of peptides[43,45−47]. A significant number of studies have employed molecular docking, bioinformatics technology, and biological protein databases to analyze the relationship between the structure and biological activity of ACE-inhibitory peptides and to elucidate the inhibitory mechanism of ACE-inhibitory peptides[48]. According to existing literature, for ACE-inhibitory peptides, peptides with smaller molecular weights have more potent biological activity than peptides with larger molecular weights[49,50]. To enhance the biological activity of the protein hydrolysate, it was subjected to a series of separation and purification processes, with the aim of aligning the final product with the specific requirements of the target market. This involved the utilization of membrane separation technology, which allowed for the separation of peptides according to their characteristics. Ultrafiltration technology has been widely used in the production of proteins and polypeptides in factories, but the size of the separation molecular weight is negatively correlated with the cost of the factory; that is, the smaller the molecular weight, the more the cost of the factory, so the factory generally chooses to use 3 kDa ultrafiltration membrane for production. Since this study will be applied to factory production, peptides < 3 kDa were selected for study.
The present study focuses on the characterization of polypeptides from the seed meal of seabuckthorn and their inhibitory activity and mechanism against ACE. Infrared spectroscopic scans, X-ray diffraction, and Quadrupole-TOF LC-MS/MS System have been used mainly for secondary and primary structure studies. The molecular weight of some active peptides identified in this study was higher than that of peptides obtained by other scholars, which may be because, for the practicality of the factory, too many separation and purification steps were not performed, and a specific peptide was not locked before identification, but the ultrafiltration samples were directly identified. In addition, the methods and databases used to identify ACE-inhibitory peptides may also lead to discrepancies and deficiencies in the results. Enzymatic products may contain short peptides not listed in the ACE inhibitor peptide database. Other reasons may be the difference in enzymes used. The enzymatic hydrolysis of food-grade enzyme preparations is weaker than that of chemical grade. Although the molecular weight of the identified peptides is slightly higher than that of other scholars, the results of previous specific research on the overall activity of ACE inhibitory peptides have shown better and can be applied in practical production.
Molecular simulation docking technology is a commonly used method to study the active mechanism of active peptides. This technology can interact with small molecules and proteins through specific algorithms and continuously optimize information such as ligand positions, amino acid side chains, and bond angles. By obtaining the binding energy of each binding mode, researchers could quickly screen out peptides with better activity based on the binding energy. This approach has extensively promoted the development of active peptide research. Li's team[48] extracted RGLSK from Pixian bean sauce, which showed vigorous ACE inhibitory activity, and Ma's team[51] extracted EYFR and LPGP from the hydrolysate of Channa striatus, which may have ACE inhibitory function. Wei's team[52] extracted seven ACE inhibitory peptides from distillers' grains, and Lu's team[53] extracted four ACE inhibitory peptides from black tea, including QTDEYGNPPR, AGFAGDDAPR, IQDKEGIPPDQQR, and SIDELR, and understood their inhibitory mechanism through molecular simulation docking technology.
This experiment uses food-grade enzymes for enzymatic hydrolysis, which avoids the distinction between chemical enzyme preparation and industrial enzyme preparation, making the experimental results more relevant to actual production and better applicable to industrial production. In this experiment, the selected peptide was not purified and synthesized, which made the ACE inhibitory peptide obtained had a higher IC50 than the peptide obtained in previous experiments, but we believe that this is in line with the needs of practical production. Moreover, the seabuckthorn seed meal peptide prepared in this study has only been tested for in vitro activity. The inhibitory effect in vivo has yet to be discovered, and further in-depth research is needed.
In this experiment, seabuckthorn seed meal protein was used as raw material, food-grade enzyme preparation was used for enzymatic hydrolysis, the yield of peptide was 24.29%, and the structure of the obtained peptide solution was characterized and the effect of ACE inhibition activity was studied. The peptide solution was ultrafiltrated, and the peptide fragment < 3 kDa was selected for structural characterization. The structure of the peptide contained in the peptide solution was mainly β-fold and β-turn structure, and it was an amorphous structure. This makes the peptide more stable and will not change in actual production. Ten peptides with ACE inhibitory activity were screened by LC-MS/MS analysis, and compared with NCBI and Uniprot databases. Molecular docking showed the conformational relationship between each peptide and ACE. The experimental analysis showed that the inhibition mode of ACE by seabuckthorn seed meal protein peptide was non-competitive. No significant effect of the inhibitory activity of the peptide occurred at different temperatures and pH. In conclusion, polypeptides from seabuckthorn seed meal showed good inhibitory stability against ACE and could be a potential drug source for the treatment of hypertension.
-
The authors confirm contribution to the paper as follows: project administration: Peng Q; formal analysis, data curation: Zheng Y, Wang D; resources, writing - review & editing: Peng Q, Yuen M, Yuen T, Yuen H; funding acquisition: Peng Q, Yuen T; validation: Zheng Y, Wang D, Yuen H; visualization: Zhou Y, Wang D; investigation: Wang D, Zhou Y, Yuen M, Yuen T; methodology: Zheng Y; writing - original draft: Zheng Y, Wang D, Zhou Y. All authors reviewed the results and approved the final version of the manuscript.
-
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
This research was financially supported by the Science and Technology Project of Xining (Grant No. 2022-Y-12) and Northwest A&F University College Students' Innovation and Entrepreneurship Training Programme (Grant No. 202410712287). The authors would also like to thank the instrument shared platform of College of Food Science & Engineering of Northwest A&F University, for the assistance in High Performance Liquid Chromatography (HPLC)/Fourier transform infrared spectrometer.
-
The authors declare that they have no conflict of interest.
- Copyright: © 2024 by the author(s). Published by Maximum Academic Press on behalf of China Agricultural University, Zhejiang University and Shenyang Agricultural University. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Zheng Y, Wang D, Zhou Y, Yuen M, Yuen T, et al. 2024. Characterization and angiotensin-converting enzyme inhibitory activity of peptides of seabuckthorn (Hippophae rhamnoides L.) seed meal. Food Innovation and Advances 3(3): 295−304 doi: 10.48130/fia-0024-0029 

Characterization and angiotensin-converting enzyme inhibitory activity of peptides of seabuckthorn (Hippophae rhamnoides L.) seed meal
- Received: 13 July 2024
- Revised: 17 September 2024
- Accepted: 20 September 2024
- Published online: 26 September 2024
Abstract: Given the side effects associated with synthetic antihypertensive drugs, there is a growing need among researchers to investigate angiotensin-converting enzyme (ACE) inhibitory peptides derived from food protein as safer therapeutic alternatives. This study used seabuckthorn (Hippophae rhamnoides L.) seed meal as the raw material, and the protein was extracted by alkaline extraction and acid precipitation. After enzymatic digestion, peptides with molecular weight less than 3 kDa were selected for study. The screened peptide had an IC50 value of 4.358 mg/mL on ACE with a non-competitive inhibition mechanism and good inhibition stability. By employing infrared (IR) analysis, exclusively β-fold and β-helix structures were identified in the hydrolysate, while no other structural motifs were detected. X-ray diffraction revealed that it had an irregular amorphous structure. The peptide contains 17 amino acids that are both highly acidic and hydrophobic, with glutamic acid ranking first in terms of the number of individual amino acids. Compared with the database (NCBI, Uniport), ten peptides with ACE inhibitory activity were detected, and molecular docking showed the mechanism of each peptide inhibiting ACE, FRVAWTEKNDGQRAPLANN, LIISVAYARVAKKLWLCNMIGDVT-TEQY, VIRSRASDGCLEVKEFEDIPP, AGGGG-GGGGGGSRRL, LQPREGPAGGTT-ALREELSLGPEAALDTPPAGP, DDEARINQLFL, FAVSTLTSYDWSDRDDATQGR-KL, RQLSLEGSGLGVEDLKDN, GGGGGGGGGGGGGGGIGGGGGGGGGGGAR, and KEALGEGCFGNRIDRIGD. According to the results, AGGGGGG-GGGGSRRL is more stable in binding to ACE and may have better inhibitory activity. It has been shown that seabuckthorn protein can be an alternative source of ACE inhibitory peptides.