








-
Clematis lanuginosa belongs to the Ranunculaceae family and is one of the most primitive and highly important parents of Clematis breeding[1]. It is also a unique wild resource in China and a key protected wild plant in Zhejiang Province and is distributed only in northeast of the province. The flowers of C. lanuginosa are usually blue–purple in colour[2,3].
Most plant flower colour is due to the presence of pigment compounds in chloroplasts or vacuoles in flower tissue. Currently known plant pigments are classified into three main categories: carotenoids, alkaloids, and flavonoids[4]. Anthocyanins constitute a large class of flavonoid compounds widely found in plant roots, stems, leaves, flowers, and fruits[5]. More than 600 anthocyanin glycosides are found in nature[6]. The most common anthocyanins in plants are six widely distributed anthocyanin glycoside derivatives: delphinidin, malvidin, pelargonidin, cyanidin, peonidin, and petunidin[7]. The type and content of anthocyanin glycosides are the main determinants of flower colour. For example, pink to purple‒red is produced by cyanidin, orange‒red to red is produced by pelargonidin, and purple to blue colouration is produced by delphinidin and its derivative malvidin pigment[8]. In addition, changes in anthocyanin content affect the final presentation of flower colour. For example, a gradual increase in cyanidin content is associated with a colour change from light to dark red[9].
Transcription factors regulate the expression intensities and patterns of structural genes associated with anthocyanin biosynthesis[10]. Wild-type petunias usually exhibit coloured stems, blue flowers, and dark brown seeds. When the coding sequence of the morning glory transcription factor InMYB1 is mutated, the stems, flowers, and seeds of the mutant plants become red, white, and white, respectively. A frameshift in the gene encoding another transcription factor, InWDR1, produces green stems, white flowers, and ivory seeds[11]. When the second intron of the TfMYB1 gene is inserted into an En/Spm-like transposon, the expression of structural genes involved in the anthocyanin synthesis pathway, such as F3H, CHS, UFGT, DFR, and ANS, is significantly downregulated, resulting in a mutant with a 'flecked' grey‒purple colour mixed with purple spots due to the significant decrease in anthocyanin content in the petals[12]. A study in peas revealed that transposon insertion into a bHLH regulatory gene inactivates this transcription factor, resulting in the formation of white flowers[13].
In C. lanuginosa, there are significant changes in flower colour across different flower development periods, and the blue–purple colour typically fades rapidly, greatly shortening the viewing period. An important goal for ornamental plant breeders is to optimize flower colour. However, there have been no reports on the biosynthesis and regulation of flavonoids in C. lanuginosa. In this study, metabolome and transcriptome analyses were used to explore colour changes during flower development in C. lanuginosa. Subsequently, key metabolites and genes were identified. The results of this study provide a direction for optimizing flower colour for higher ornamental value.
-
Plant materials were obtained from C. lanuginosa plants with consistent flower colour planted in the Clematis germplasm resource garden of the Zhejiang Institute of Subtropical Crops (120°63'54" E, 27°99'88" N), Zhejiang, China. The flower colours at the six selected stages were analysed and described according to the Royal Horticultural Society Colour Chart (RHSCC). Under the same lighting conditions, the middle part of the flower was compared to the RHSCC card. The RHSCC value of a flower is indicated by the code closest to the colour on the RHSCC card, and the colour range and number to which it belongs were determined[14,15]. After flower colour determination, flowers at the six stages were collected three times for subsequent research.
Metabolite extraction and analysis
-
The C. lanuginosa flower samples were processed as follows: first, the flowers were vacuum freeze-dried and then powdered in a ball mill at 30 Hz for 15 min; then, 50 mg of the powder was weighed, dissolved in 500 μL of extraction solution (50% methanol aqueous solution containing 0.1% hydrochloric acid), vortexed for 5 min, sonicated for 5 min, centrifuged at 12,000 r/min and 4 °C for 3 min, and pipetted twice after centrifugation. The supernatant was pooled, filtered through a 0.22 μm microporous membrane, and stored in an injection vial for subsequent LC‒MS/MS analysis[16,17].
MultiQuant 3.0.3 software was used to process the raw data acquired by tandem mass spectrometry (MS/MS) and ultra-performance liquid chromatography (UPLC) (ExionLC™ AD), including reference standard retention time and peak shape information. The metabolites in flowers of different colours were analysed by integrating the mass spectral peaks of the analytes in different samples to ensure the accuracy of the qualitative quantification, followed by cluster analysis and principal component analysis[18].
The differentially abundant metabolites were identified based on the difference multiplier value (fold_change) and the p value obtained using the Wilcoxon rank sum test method[19] or t-test (or based on the difference factor value (fold_change) alone when there were no biological replicates). Metabolites with fold changes ≥ 2 and ≤ 0.5 were selected as the final differentially abundant metabolites. A clustering correlation heatmap with signs was generated using OmicStudio tools (
www.omicstudio.cn ).Full-length transcriptome sequencing and data analysis
-
Eighteen RNA samples (three biological replicates of each sample) were mixed in equal amounts. Single-molecule real-time (SMRT) library construction was performed as follows: oligo (dT)-enriched mRNA containing poly-A was generated and then reverse transcribed to cDNA using a SMARTer PCR cDNA Synthesis Kit and enriched cDNA was amplified by PCR. A sample of the cDNA was screened by BluePippin, and fragments larger than 4 kb were then enriched. Screened fragments were amplified by large-scale PCR to obtain sufficient cDNA and then subjected to damage repair, end repair, and ligation of the SMRT dumbbell-type linker. Nonscreened fragments were then mixed at an equimolar ratio with fragments larger than 4 kb to construct the final library.
Exonuclease digestion was performed, and the unligated junctions at both ends of the cDNA sequence were then removed. The primers and DNA-binding polymerase were combined to generate a complete SMRTbell library. After the library was qualitatively analysed, the PacBio Sequel platform was used for sequencing according to the effective concentration of the library and the data output requirements. The official SMRT Link v6.0 software package was used to filter and process the data, and circular consensus sequence (CCS) data containing full-length and non-full-length fragments were generated. The full-length nonchimeric (FLNC) sequence and nonfull-length (nFL) nonchimeric sequence were subsequently used to determine whether the CCS contained the 5'-primer, 3'-primer, and poly-A sequences. The isoform-level clustering (ICE) algorithm was used to cluster the FLNC sequences of the same transcript to obtain the consensus sequence. The nFL nonchimeric sequences were subsequently used to correct the obtained consensus sequence, and the polished consensus sequence was ultimately obtained. The NGS data were used to correct the polished consensus sequence using LoREDC software.
De novo sequencing of different flower stages and data analysis
-
Total RNA from eighteen C. lanuginosa flower samples (three biological replicates of each sample) was used to construct an mRNA library and high-throughput sequencing was performed using an Illumina HiSeq™ 2500 sequencer. Paired-end 150 bp data were obtained and de novo processed into transcripts and unigenes as reference sequences for subsequent analysis[20].
The assembled unigenes were annotated using six databases (NR, SwissProt, Pfam, COG, GO, and KEGG). The BLAST2GO program performs GO classification and then maps unigenes to the KEGG database to determine their associated metabolic pathways. The quantitative expression results were then used for differential gene analysis between groups to obtain the differentially expressed genes; the difference analysis software used was DESeq2, with a screening threshold of |log2FC| > 1 and adjusted p < 0.05.
Real-time quantitative (qRT‒PCR) validation and analysis
-
Real-time PCR primers were designed using Oligo 7 software. GAPDH was used as the internal reference gene. The primers used are shown in Table 1. The reaction system used was described in a previous study by Ye et al.[21]. Three technical replicates were performed to ensure the accuracy of the experiment, and the relative expression was calculated using the 2−ΔΔCᴛ method.
Table 1. Primers for qRT-PCR.
Gene Primer-F Primer-R GAPDH AACCCCTGAGGAGATTCCA CACCACCCTTCAAGTGAGCAG ANS1 ATTGTGCACATCGGTGACAC CGACTCACTGACAAGTTCTG ANS2 TGCCTGGTCTCCAAGTGTAC CTAGCCCTCTATGCAGTATAC F3'H1 TCTTGTTGAGTACATCTTGG GACACTAGGTGGCAAGCGTG WD40 ATGAGCGAGAATTGCTGAGC TGCTACTGTGCATCCATCTG MYB1 AAGGCCGTTGGGATACGTTA ATCCTAGACCACCTGTTGCC MYB2 CACTGTTACCTCCGACGAGA CAGGTCTGTATCCTCGCTGT bHLH1 TGAAGACACCTGAAGGGCAA TCGTTGGAGCAAGATTCGGT bHLH2 TGCGAAGGAGTTCTGGTGAA ATGGCAAGAGAAGTCCCGAA Combined metabolome and transcriptome analysis to explore anthocyanin production mechanisms
-
Combined analysis of the transcriptome and metabolomic data was performed using WGCNA with default parameters in R to simplify the gene expression data into coexpressed modules, normalize the FPKM values, and construct adjacency matrices. The phenotypic data were imported into the WGCNA package, and correlation-based associations between phenotypes and gene modules were identified using default settings. The WGCNA package was used to convert adjacency matrices into topological overlap matrices (TOMs)[22]. After network construction, transcripts with the same expression pattern were grouped to establish modules, and feature calculations were performed. Cytoscape 3.9.1 with default parameters was used to draw the network diagram[23].
-
The stages of development of C. lanuginosa flowers were analysed with a colorimetric card. The flower colours at the six stages are described as follows (Table 2): yellow‒green (bud stage, myfs1, N144D), light blue‒purple (colouring stage, myfs2, N88C), blue‒purple (early flowering stage, myfs3, N88B), bright blue‒purple (flowering stage, myfs4, 90D), lilac blue‒purple (after flowering stage, myfs5, 91B), and extremely lilac purple (end flowering stage, myfs6, 91C) (Table 2 & Fig. 1).
Table 2. Flower color identification of C. lanuginosa.
Sequencing number Flower stage RSHCC Colour myfs1 Bud stage N144D yellow‒green myfs2 Coloration stage N88C light blue‒purple myfs3 Early flowering stage N88B dark purple myfs4 Flowering stage 90D bright blue‒purple myfs5 Post flowering stage 91B lilac purple stage myfs6 End flowering stage 91C extremely lilac purple stage 
Figure 1.
Different stages of C. lanuginosa flower development. (a) Bud stage (myfs1, yellow‒green, N144D); (b) Colouring stage (myfs2, light blue‒purple, light blue‒purple, N88C); (c) Early flowering stage (myfs3, dark purple, N88B); (d) Flowering stage (myfs4, bright blue‒purple, 90D); (e) After the flowering stage (myfs5, lilac purple stage, 91B); (f) End flowering stage (myfs6, extremely lilac purple stage, 91C).
Metabolomics assays
-
Visual examination of the total ion current (TIC) plots revealed a strong instrumental analysis signal, a large peak capacity, and good retention time reproducibility for all the samples (Fig. 2a). The essential metabolite compositions in the different flowers of C. lanuginosa were determined by gas chromatography–mass spectrometry (GC‒MS), and this analysis identified 25 compounds in the flowers. Overall, the number of species and quantity of primary metabolites were greater than those of secondary metabolites, indicating that the flower samples presented vigorous primary metabolic activities. For these metabolites, principal component analysis (PCA) accurately grouped all the samples into distinct clusters, which reflected the obvious differences between the different flower stages (Fig. 2b).
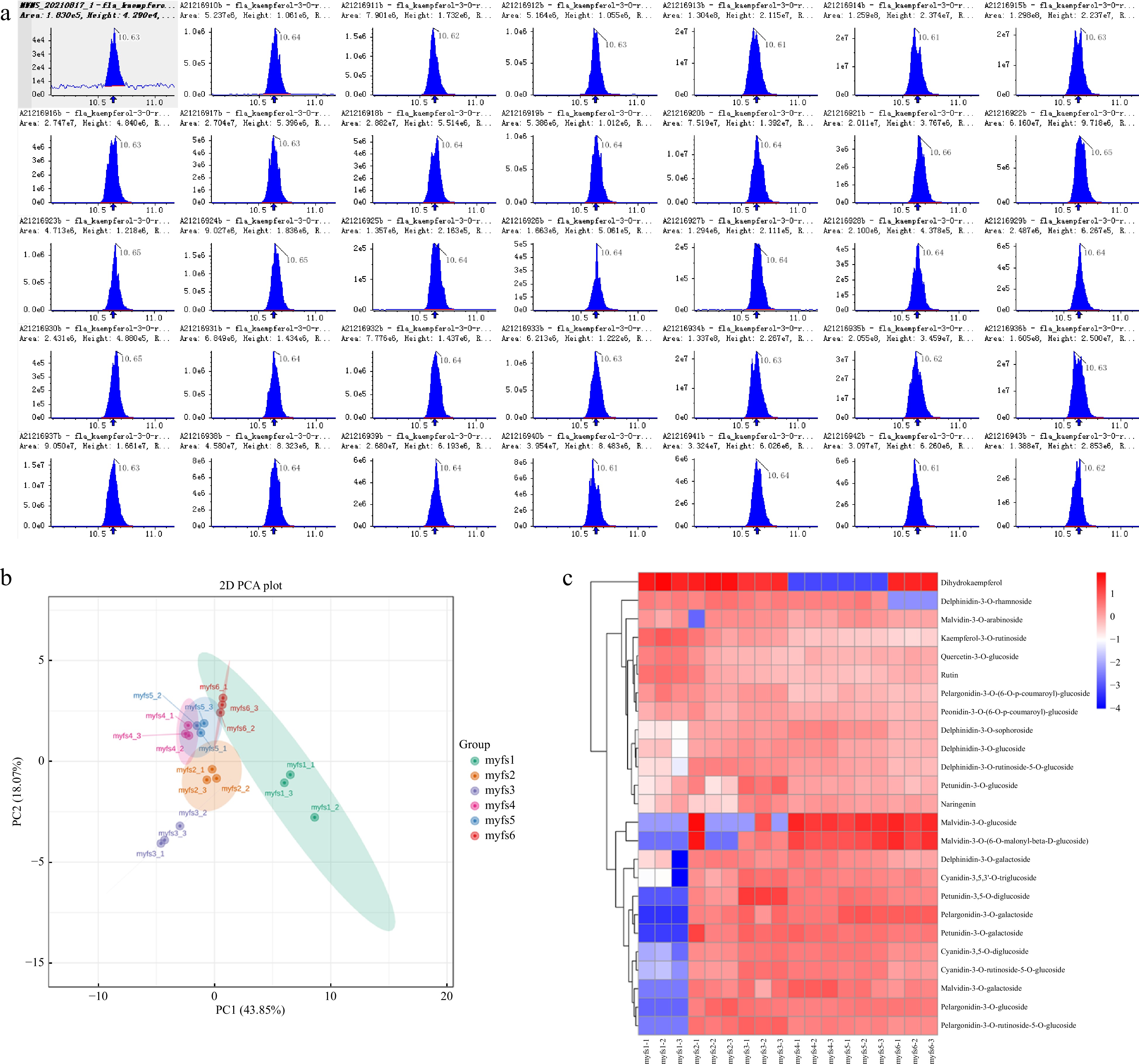
Figure 2.
Metabolomic analysis of the different flowers of C. lanuginosa. (a) Total ion current (TIC) plots of all the samples. (b) Principal component analysis (PCA) plots. Different groups are represented by different colours. (c) Heatmap of metabolites. Different groups are represented by different colours. The X-axis shows the different samples, and the Y-axis shows the metabolites in the different flowers of C. lanuginosa. The upregulated and downregulated genes are shown in red and blue, respectively.
All the detected metabolite content data were normalized, and heatmaps were generated (Fig. 2c). The results revealed that cyanidin-3-O-galactoside and cyanidin-3-O-ruticoside-5-O-glucoside were expressed only in the myfs1 stage. The expression levels of cyanidin-3,5,3'-O-triglucosidecyanidin-3,5-O-diglucoside, cyanidin-3-O-ruticoside-5-O-glucoside, pelargonidin-3-O-rutin-5-O-glucoside, petunidin-3,5-O-diglucoside, petunidin-3-O-glucoside, and delphinidin-3-O-glucoside tended to increase during the first three developmental stages, peaked during the myfs3 period, and then tended to decrease.
In addition, 22 differentially expressed metabolites were identified (Fig. 3a). The most differentially abundant metabolites were observed between myfs3 and myfs6. To further evaluate the specific expression profiles of the differentially expressed metabolites, we normalized the expression data of the differentially expressed metabolites were normalized and a heatmap generated (Fig. 3b). Petunidin 3,5-O-diglucoside, petunidin 3-O-glucoside, pelargonidin-3-O-ruticoside-5-O-glucoside, cyanidin-3,5,3'-O-triglucoside, cyanidin-3,5-O-diglucoside, and cyanidin-3-O-ruticoside-5-O-glucoside were expressed during the myfs1 period and then tended to increase, peaking in the early flowering period (myfs3), and then gradually decreasing.
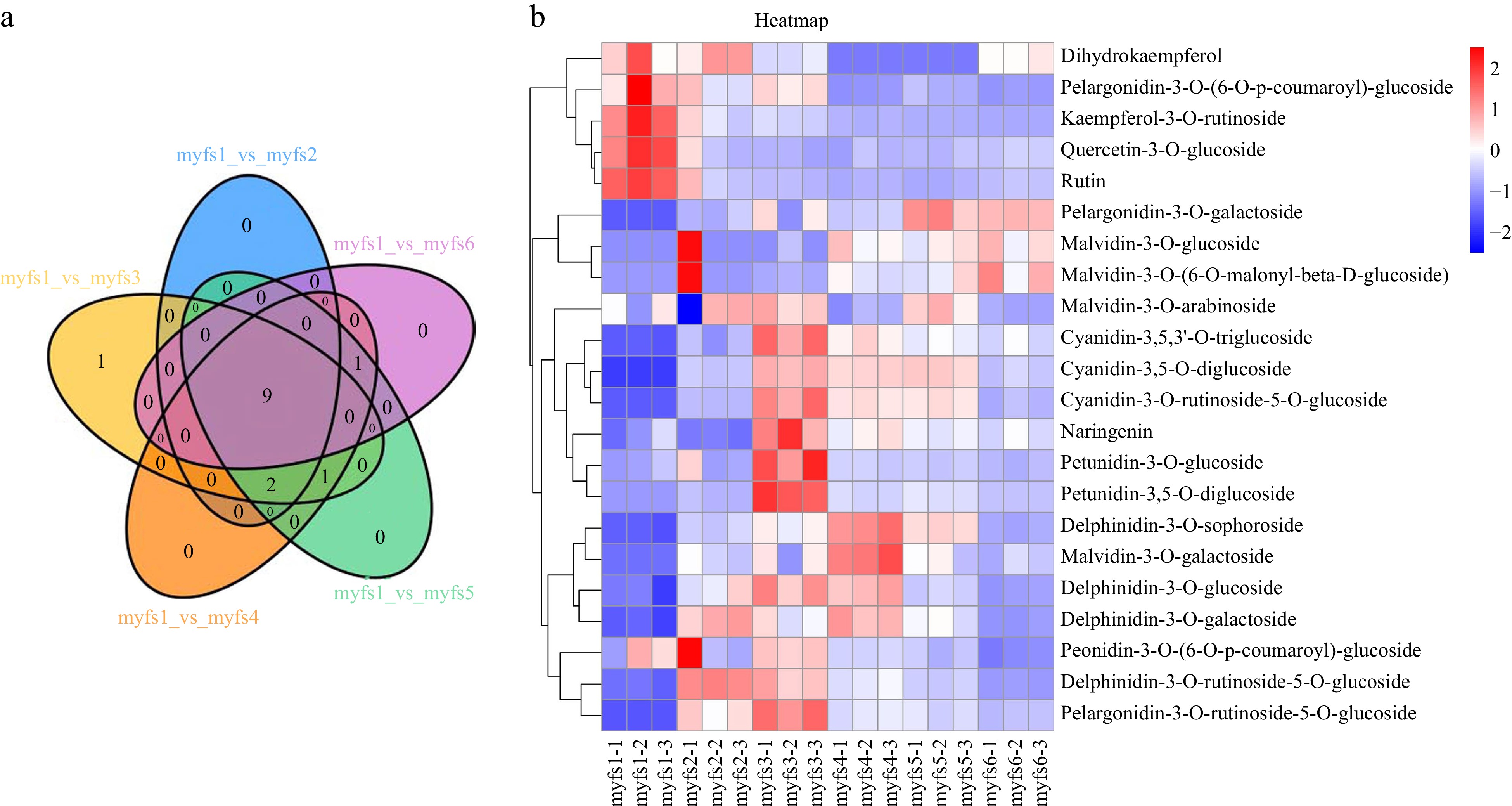
Figure 3.
Analysis of differentially expressed metabolites in flowers of C. lanuginosa. (a) Venn diagram of differentially expressed metabolites. Different comparison groups are represented by different colours. (b) Heatmap of differentially expressed metabolites. Different groups are represented by different colours. The X-axis shows the different samples, and the Y-axis shows the metabolites. The upregulated and downregulated genes are shown in red and blue, respectively.
Transcriptome sequencing data analysis
-
A total of 773,680 CCS and 643,906 full-length nonchimeric (FLNC) sequence reads were obtained via full-length transcriptome sequencing. The FLNC cluster-corrected sequence was corrected using data from Illumina deep sequencing, and 54,793 gene sequences were obtained by removing redundant and similar sequences from the sequences obtained using CD-HIT software, with an average length of 2,365 bp and an N50 of 3,088. Gene function annotations in seven databases, namely, Nr, KEGG, Nt, Pfam, KOG/COG, SwissProt, and GO, revealed a total of 20,896 transcripts annotated; the most genes (17,656) were annotated in the SwissProt database, whereas the fewest genes were annotated in the GO and Pfam databases (13,377 each), and 7,416 genes were annotated in all seven databases (Fig. 4a). Using ANGEL software to predict the CDSs of the full-length transcriptome[24], 25,800 CDSs were obtained, of which 20.27% (5,230) had CDSs with lengths greater than 1,800 bp (Fig. 4b).
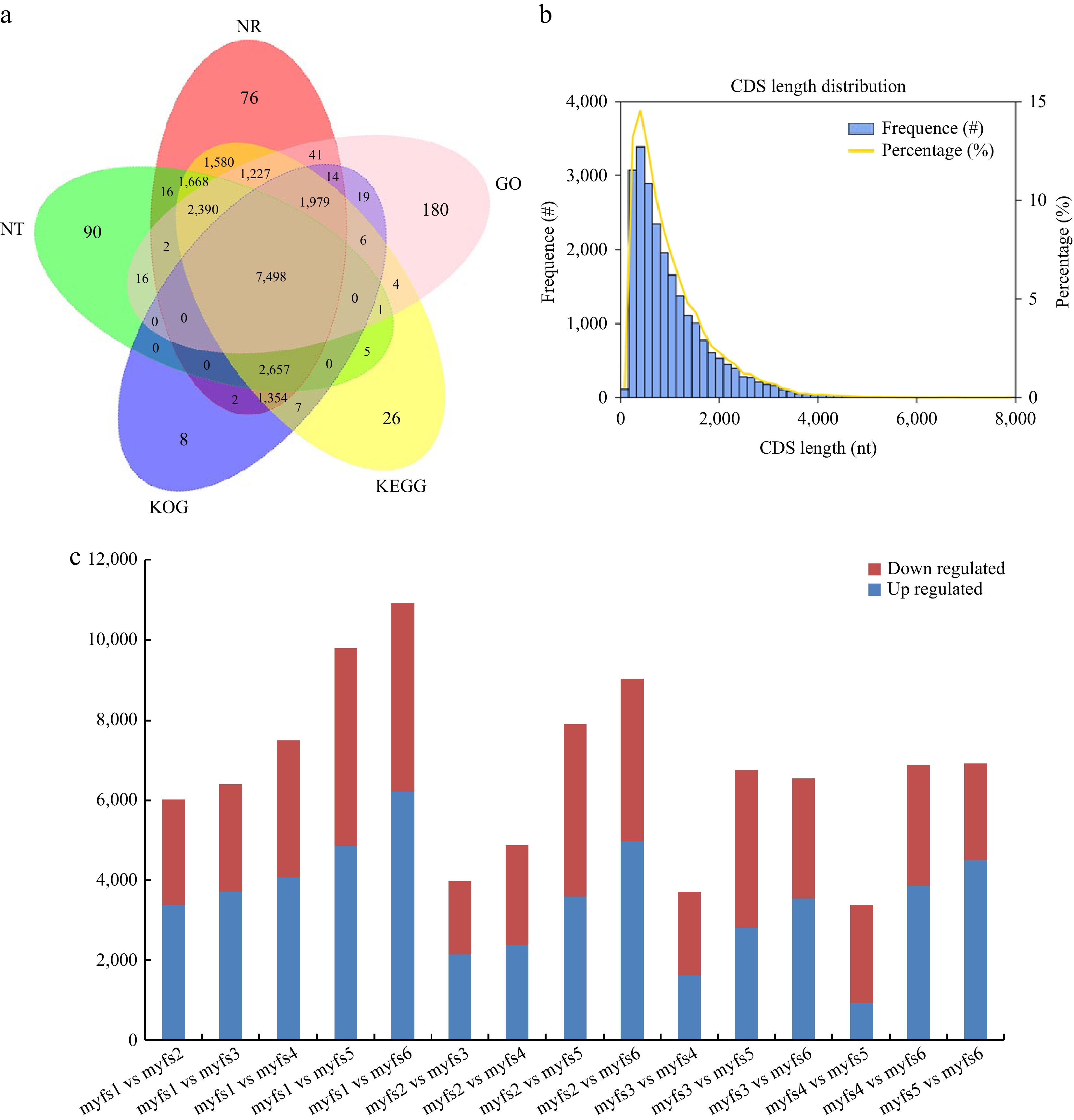
Figure 4.
Transcript annotation and CDS prediction. (a) Venn diagram of the full-length transcriptome annotation results; different comparison groups are represented by different colours. (b) CDS prediction in the full-length transcriptome. The X-axis shows the CDS length, the left Y-axis shows the occurrence frequency of the CDSs of different lengths, and the right Y-axis shows the proportion of CDSs of different lengths. (c) DEGs during C. lanuginosa flower development. Red bars represent upregulated DEGs, and blue bars represent downregulated DEGs.
To explore the molecular mechanisms involved in the development of C. lanuginosa flowers, different flower developmental stages of C. lanuginosa were analysed by next-generation transcriptome sequencing. From these sequences, 169.42 GB of clean data were obtained, with a filtering error rate of less than 0.025%, a Q30 between 94.6% and 95.17%, and a GC content between 45% and 45.77%. To explore the DEGs among C. lanuginosa flowers at the six different stages, the FPKM values of all genes in the samples were compared. The greatest number of DEGs was found between myfs1 and myfs6 (10,909; 6,216 upregulated and 4,693 downregulated), followed by myfs1 vs myfs5, and myfs2 vs myfs6. The smallest number of DEGs was found between myfs4 and myfs5, with a total of 3,382 DEGs (936 upregulated and 2,446 downregulated), followed by myfs3 vs myfs4, and myfs2 vs myfs3 (Fig. 5).
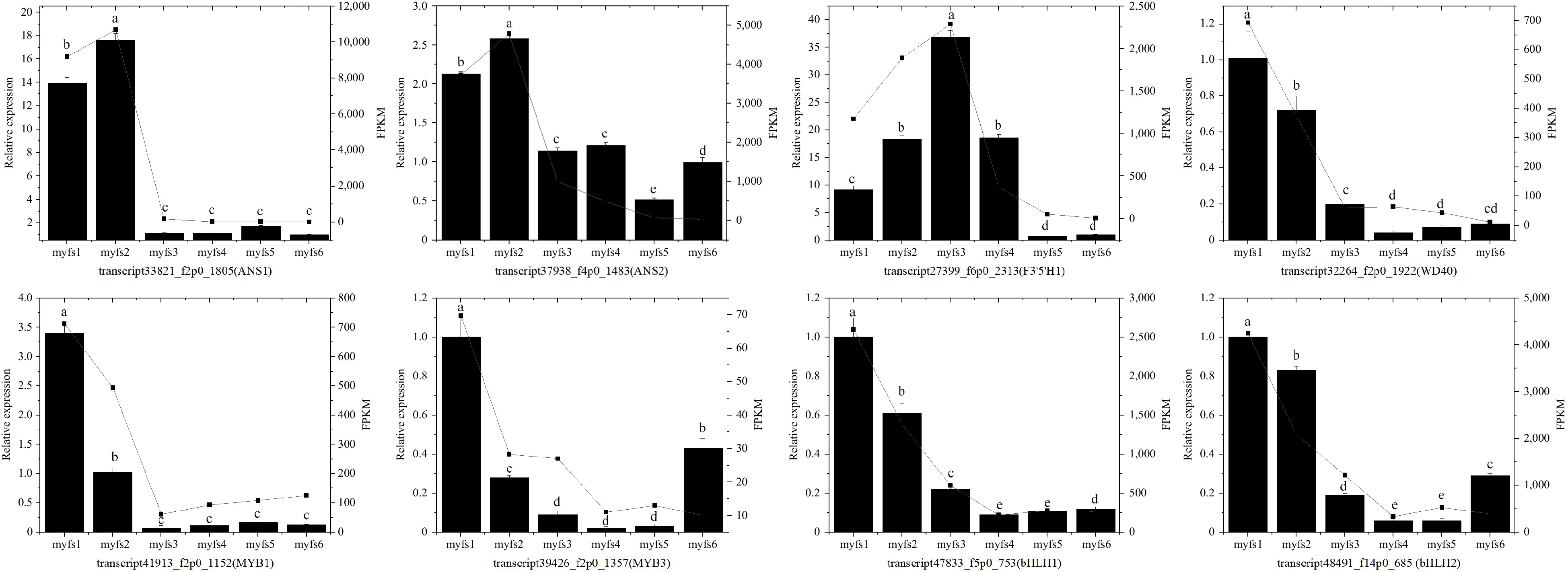
Figure 5.
Identification of the expression profiles of eight unigenes in C. lanuginosa. The bar chart presents the relative expression levels of the genes, and the point line diagram presents the FPKM values of the unigenes. The left Y-axis represents the relative expression, whereas the right Y-axis represents the FPKM values of the unigenes. (a)−(e) Indicate statistical significance.
Real-time PCR validation
-
To further verify the reliability of the transcriptome sequencing results, eight unigenes (two ANS genes, two MYB genes, two bHLH genes, one F3'5'H gene, and one WD40 gene) were randomly selected for expression-level detection by qRT–PCR. As shown in Fig. 5, the expression trends of the qRT‒PCR results and the FPKM values from the sequencing results are consistent, suggesting that the transcriptome results and analysis are reliable and can be used for further analysis (Fig. 5).
Combined metabolomic and transcriptomic analyses resolve anthocyanin synthesis
-
For the WGCNA of the FPKM values, myfs1 was used as the control group, and 27 gene modules were identified according to the coexpression patterns of individual genes. These gene modules are represented in different colours in cluster maps and network heatmaps (Fig. 6a). Using different metabolites at different stages of C. lanuginosa development as phenotypic data, module‒trait correlations were analysed, and a sample tree map and a trait heatmap were constructed to clarify the expression of metabolite contents at different developmental stages (Fig. 6b).
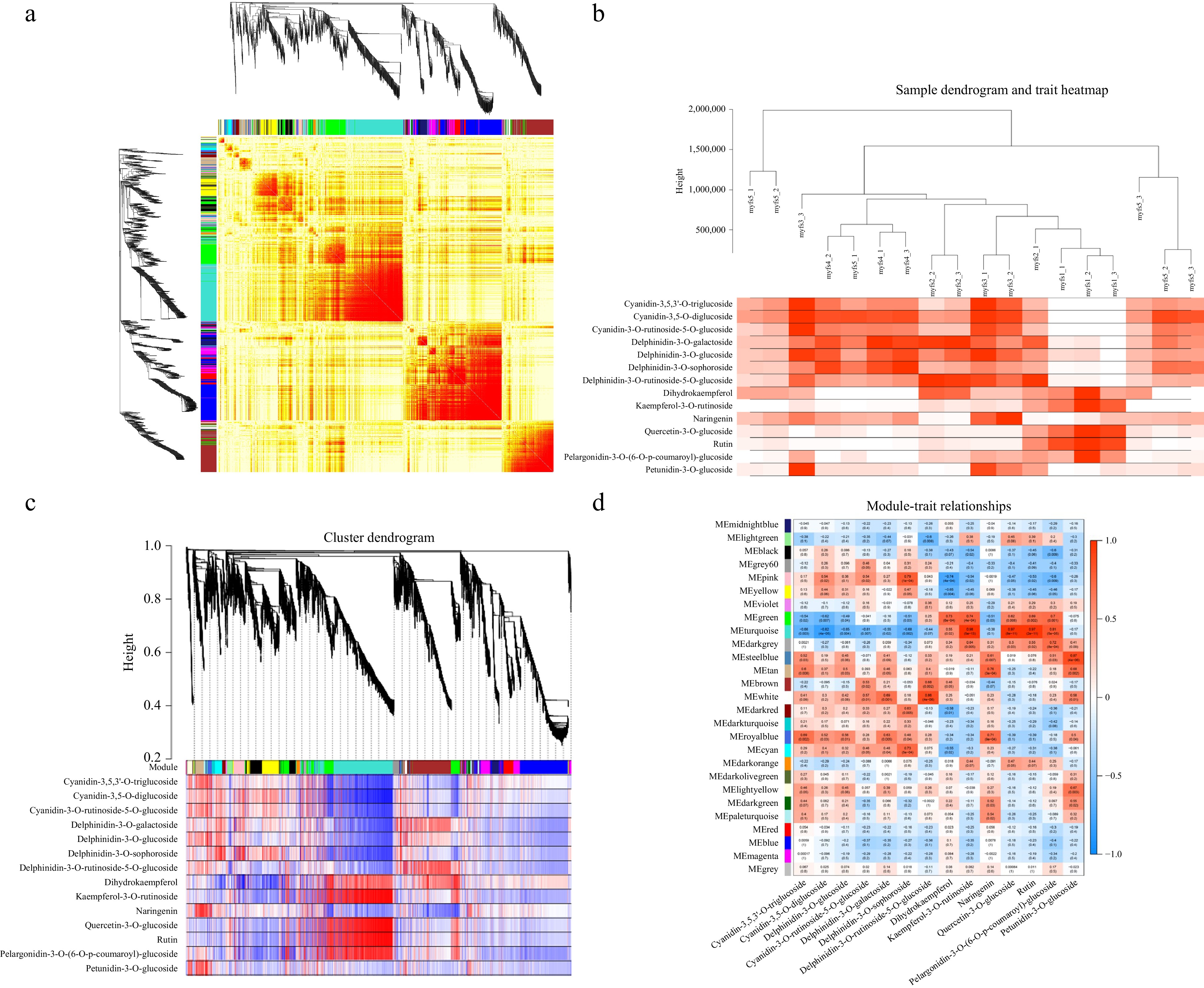
Figure 6.
Coexpression patterns of metabolism-related genes. (a) Cluster dendrogram and network heatmap of the genes calculated by the coexpression module. (b) Modular trait heatmap of the sample dendrograms at each developmental stage. (c) Hierarchical clustering showing the 11 modules with coexpressed genes. (d) Module‒trait associations based on Pearson correlation. The colours from green to red represent −1 to1.
One of these 27 gene modules showed a very significant relationship with the differentially expressed metabolites. The MEroyalblue gene module is associated with anthocyanin biosynthesis, with the highest correlations with cyanidin-3,5-O-diglucoside, cyanidin-3-O-rhamnoside glycoside-5-O-glucoside, and cyanidin-3,5,3'-O-triglucoside, with R2 values of 0.69, 0.52, and 0.56, respectively (Fig. 6c & d).
Correlation analysis of the differentially expressed MYB, bHLH, and WD40 transcription factors and differentially abundant metabolites in all the gene modules revealed that the MYB gene (transcript41913_f2p0_1152), cyanidin-3,5-O-diglucoside, cyanidin-3-O-rutin-5-O-glucoside, and cyanidin-3,5,3'-O-tri glucoside had the highest correlations, suggesting that this MYB gene may be a key gene involved in colour formation in C. lanuginosa (Fig. 7).
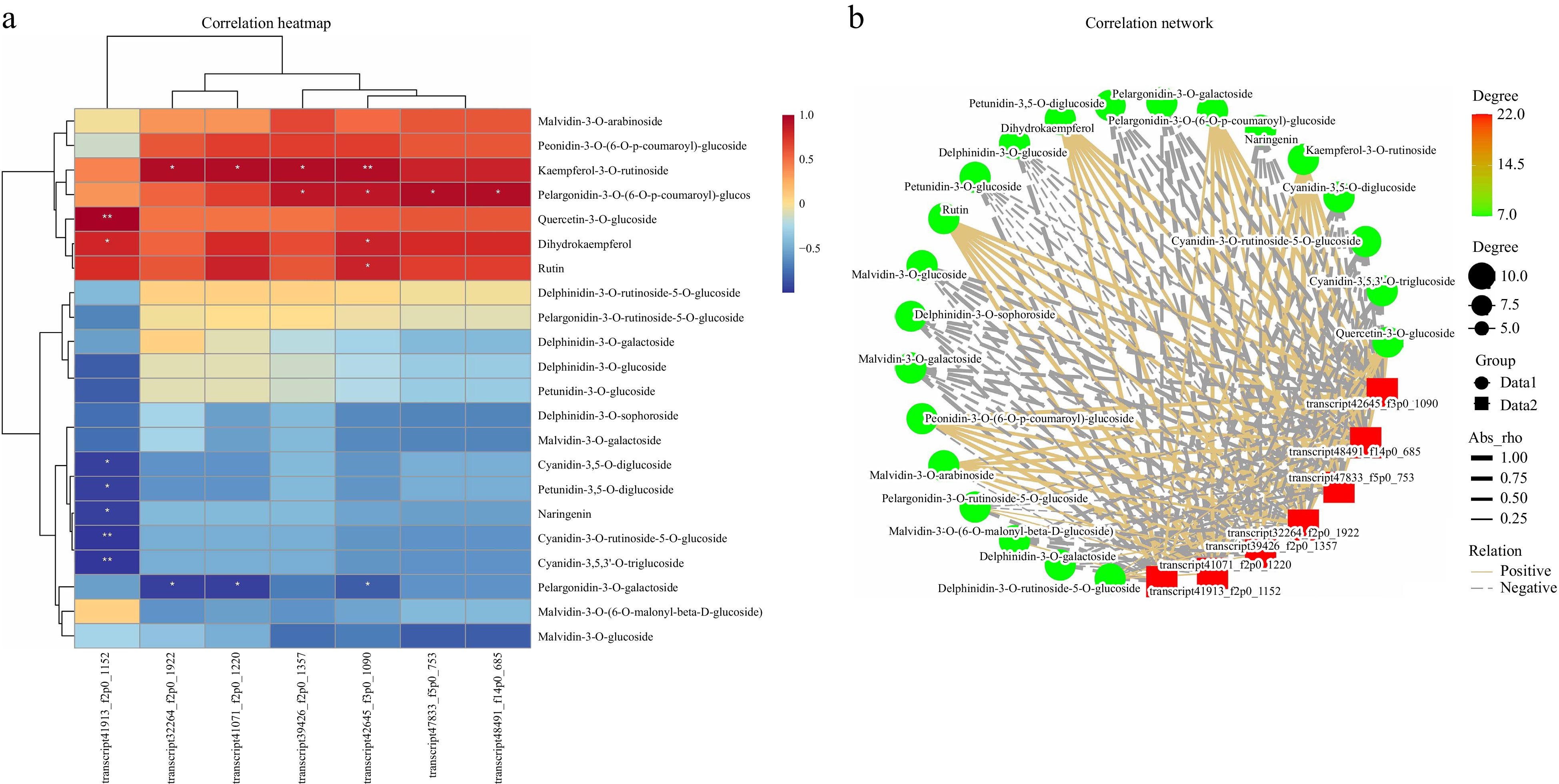
Figure 7.
Correlation analysis of transcription factors and differentially expressed metabolites. (a) Heatmap of transcription factors and differentially expressed metabolites; the X-axis represents transcription factors, and the Y-axis represents differentially expressed metabolites. (b) Network plots of transcription factors and differentially expressed metabolites. Red represents transcription factors, and green circles represent differentially expressed metabolites.
-
Flower characteristics are important traits of many ornamental plants[25]; this is also true for C. lanuginosa (widely used in home gardening and urban greening). In C. lanuginosa, during the early flowering period (myfs3), the flowers are blue–purple, which is a rare colour. Delphinidin is the first type of anthocyanin that accumulates in the pigment synthesis pathway of blue–violet flowers and is the most important basal pigment[26]. Malvidin and petunidin, which represent different degrees of delphinidin methylation, are also important colour-forming substances for many purple plants[27,28]. Mizuta suggested that blue anthocyanins are dominated by delphinidin, malvidin, and petunidin[29]. In this study, petunidin-3,5-O-diglucoside, petunidin-3-O-glucoside, and delphinidin-3-O-glucoside expression levels began to increase at the myfs1 stage and reached their highest levels during the early flowering period (myfs3). These results suggest that these three metabolites may be involved in the development of flower colour during the early flowering period, resulting in a blue‒purple colour.
However, the flowers of C. lanuginosa in the myfs3 stage are not only pure blue‒purple but also purple‒red. Previous reports have indicated that cyanidin produces a magenta flower colour, whereas pelargonidin produces a brick-red flower colour[30,31]. In this study, cyanidin-3,5,3'-O-triglucoside, cyanidin-3,5-O-diglucoside, cyanidin-3-O-rutinoside-5-O-glucoside, and pelargonidin-3-O-rutinoside-5-O-glucoside were upregulated in the myfs3 stage, indicating that they may be important compounds for flower colour formation in C. lanuginosa and, in combination with the abovementioned metabolites, produce blue–purple flowers.
In addition, in the end flower development stage (myfs6), cyanidin-3,5,3'-O-triglucoside, cyanidin-3-O-rutinoside-5-O-glucoside, delphinidin-3-O-glucoside, pelargonidin-3-O-rutinoside-5-O-glucoside, petunidin-3,5-O-diglucoside and petunidin-3-O-glucoside were significantly downregulated. The loss of these metabolites may cause the colour of C. lanuginosa to become lighter, as the flower colour gradually becomes white in later stages[32].
Candidate transcription factors involved in anthocyanin biosynthesis
-
Flowering time is an important life history trait in plants and is regulated by both internal and environmental factors[33,34]. Transcription factors mainly regulate the transcription of structural genes and thus participate in anthocyanin biosynthesis. By binding to cis-acting elements in the promoters of structural genes, transcription factors activate or inhibit the expression of one or more structural genes in the anthocyanin biosynthesis pathway, and functional proteins can coordinate these interactions[35,36]. Jin et al. reported that the MYB transcription factor PavMYB10.1 participates in anthocyanin biosynthesis in sweet cherry, thereby affecting fruit colour[37]. Notably, MdMYB10 participates in anthocyanin synthesis and regulates fruit colour[38]. Stracke et al. reported that in the model plant Arabidopsis, the transcription factor MYB can regulate the expression of early structural genes such as FLS, CHI, F3H, and CHS[39].
In addition, previous studies have reported that ternary protein complexes formed by the MYB, bHLH, and WD40 genes play important roles in regulating anthocyanin biosynthesis[40,41]. In Arabidopsis, the MBW transcription complex WD40 was found to interact with different MYB transcription factors[42]. In the MBW ternary complex, MYB and bHLH transcription factor interactions are prerequisites for the recognition of specific DNA sequences. Moreover, WD40 transcription factors can increase the stability of ternary complexes[43]. Therefore, the MBW ternary complex, like other transcription factors, acts mainly by regulating the transcriptional abundance of structural genes, and simultaneously, each member can also coordinate with the others[44]. In a study in Arabidopsis, Li reported that the MYB–bHLH–WD40 (MBW) complex activated late anthocyanin biosynthetic genes[45]. Zhao et al. reported that FaMYB9/FaMYB11, FabHLH3, and FaTTG1 are functional homologues of AtTT2, AtTT8, and AtTTG1 in strawberry and promote strawberry fruit growth[42]. Feller et al. reported that the interaction between bHLH and R2R3-MYB proteins plays an important role in the colour production process of maize[46]. In this study, 15 differentially expressed MYB genes, eight bHLH genes, and 26 WD40 genes were identified, and relevant transcription factors were also predicted. Changes in the anthocyanin content of C. lanuginosa flowers may be related to and caused by these transcription factors. Specifically, the combined metabolic‒transcriptional analysis suggested that the MYB gene (transcript41913_f2p0_1152) may be a key gene involved in the development of C. lanuginosa flower colour.
-
To further study colour development in C. lanuginosa flowers, in this study, metabolome and transcriptome sequencing was performed at six flower development stages (myfs1, myfs2, myfs3, myfs4, myfs5, and myfs6). The metabolome sequencing results revealed 25 anthocyanin compounds, including 22 differentially expressed metabolites. Cyanidin-3,5-O-diglucoside, cyanidin-3,5,3'-O-triglucoside, cyanidin-3-O-rutinoside-5-O-glucoside, delphinidin-3-O-glucoside, and petunidin-3-O-glucoside may affect the formation of blue–purple flowers in C. lanuginosa. The combined metabolome and transcriptome analysis results revealed that the MYB gene (transcript41913_f2p0_1152) is a key gene involved in C. lanuginosa flower colour development and change.
This research was supported by the National Natural Science Foundation of China (32102428); the Zhejiang Science and Technology Major Program on Agricultural New Variety Breeding (2021C02071-6); Wenzhou Agricultural New Variety Breeding Cooperative Group Project (ZX2024004-3).
-
The authors confirm contribution to the paper as follows: experiments design: Ye Y, Qian R; experiments performing: Ye Y, Qian R, Hu Q, Ma X; manuscript preparation: Ye Y, Qian R, Gao H, Zheng J. All authors reviewed the results and approved the final version of the manuscript.
-
The data produced in the study were uploaded to the NCBI database (BioProject: PRJNA1117522, SRA: SRP510351).
-
The authors declare that they have no conflict of interest.
-
# Authors contributed equally: Renjuan Qian, Youju Ye
- Copyright: © 2025 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Qian R, Ye Y, Ma X, Gao H, Hu Q, et al. 2025. Targeted metabolome and transcriptome analysis reveals the key metabolites and genes influencing blue–purple colour development in Clematis lanuginosa flowers. Ornamental Plant Research 5: e001 doi: 10.48130/opr-0024-0031 

Targeted metabolome and transcriptome analysis reveals the key metabolites and genes influencing blue–purple colour development in Clematis lanuginosa flowers
- Received: 24 February 2024
- Revised: 17 October 2024
- Accepted: 11 November 2024
- Published online: 09 January 2025
Abstract: Clematis lanuginosa, a valuable ornamental plant in Zhejiang Province, China, produces flowers that are blue–purple, a rare flower colour. In this study, to explore the anthocyanin synthesis mechanism involved in flower colour formation in C. lanuginosa, metabolome, and transcriptome sequencing was performed at six flower development stages. Metabolome analysis revealed 25 anthocyanin compounds and 22 differentially expressed metabolites. Cyanidin-3,5-O-diglucoside, cyanidin-3,5,3'-O-triglucoside, cyanidin-3-O-rutinoside-5-O-glucoside, delphinidin-3-O-glucoside, and petunidin-3-O-glucoside may promote the formation of blue–purple colour in flowers. The combined analysis results revealed that the transcript41913_f2p0_1152 gene (MYB-like) may be a key gene in C. lanuginosa blue–purple flower colour development. These results provide the basis for further research on the blue–purple flower colour of C. lanuginosa.
-
Key words:
- Clematis lanuginosa /
- Flower colour /
- Blue–purple /
- Combined analysis