










-
Tea aroma is affected by the tea cultivars, growing environment, and manufacturing process, and is an essential factor in determining the quality of tea[1]. As a high-weight trait in tea plant breeding, the volatile content of tea cultivars with different adaptability is usually different[2,3]. Studies have shown that the contents and ratios of linalool and geraniol are genetically specific and stable in tea cultivars[4]. The aroma quality of oolong tea is related to the release of aroma glycosides in the leaves of cultivars during the shaking process[5]. The major volatiles in tea are derived from either the terpenoid and shikimate pathways or by the oxidation of fatty acids and carotenoids[6]. It was found that aroma components can be synthesized by a single gene or multiple gene interactions[7]. Moreover, many transcription factor (TFs) can also participate in the formation of volatiles by regulating the expression of aroma synthesis pathway genes[8]. In summary, the formation and regulation of tea aroma is a complex process.
Oolong tea, which is a semifermented tea, possesses an elegant floral odour and is gaining popularity in China due to its distinct and characteristic aromas. Furthermore, the aroma quality of oolong tea can vary greatly because of cultivars, tea manufacturing process, regions, climate conditions, season of harvest, and quality of fresh tea leaves, with the cultivar being the most important factor[2, 3]. 'Tieguanyin' (TGY, Registration No. GS13007-1985) tea is a typical cultivar of Chinese oolong tea, which is famous for its unique rich flavour and orchid-like aroma[2, 9, 10]. 'Jinxuan' (JX, Registration No. MS2011002) is one of the main cultivated tea cultivars in Fujian Province, China. Oolong tea processed from JX is popular among tea drinkers due to its unique floral and creamy aroma[11]. 'Fujian Shuixian' (FJSX, Registration No. GS13009-1985) is considered to be one of the most suitable cultivars for producing oolong tea[12]. Oolong tea processed from 'Shuixian' is popular among tea drinkers due to its sharp and typical floral odour[13]. White tea is a lightly fermented tea popular for its sweetness, clear fragrance, mellow aroma and outstanding health benefits, and it is rich in volatile compounds inherent to fresh leaves, such as aldehydes and alcohols[14, 15]. 'Fuding Dabaicha' (FDDB, Registration No. GS13001-1985) is a major cultivar suitable for making white tea and an important parent for breeding green and black tea cultivars, which played important roles in the Chinese tea breeding history[16]. Chinese green tea, the most popular tea in China, presents different characteristic aroma types according to its sensory quality, such as floral, green, and delicate aromas[17]. For instance, the representative green tea cultivars 'Longjing43' (LJ43, Registration No. GS 13037-1987), 'Baihaozao' (BHZ, Registration No. GS13017-1994), and 'Shuchazao' (SCZ, Registration No GS2002008), which come from Fujian, Zhejiang, Hunan, and Anhui Provinces, respectively, were identified as Chinese national improved cultivars.
In our previous study[18], we have analyzed the characteristic metabolites of seven tea cultivars using targeted metabolomics and widely targeted metabolomics, and combined transcriptome data to construct transcriptional regulatory networks for the characteristic metabolites of different cultivars. In addition to non-volatile metabolites, tea cultivars also affect the content of aromatic substances in its fresh leaves, which are the material basis for the formation of tea aroma. Although there have been many studies on the volatile components of tea, most of them have focused on tea processing and finished tea[19−21], and the influence of tea cultivars on aroma formation has received little attention. In this study, volatile metabolomics and transcriptomics were used to analyze the characteristic aroma components and differential genes of seven tea cultivars. Then the transcriptome data and aroma components were correlated by weighted gene co-expression network analysis (WGCNA), and co-expressed gene modules were screened to construct transcriptional regulatory networks of characteristic aroma components. These data and results will provide a theoretical basis for the production adaptability of tea cultivars at the aroma component and molecular level.
-
In April 2021, the young shoots (one bud and two leaves) of the tea plants of Camellia sinensis (L.) O. Kuntze 'Tieguanyin' (TGY), 'Jinxuan' (JX), 'Fujian Shuixian' (FJSX), 'Fuding Dabaicha' (FDDB), 'Baihaozao' (BHZ), 'Longjing 43' (LJ43), and 'Shuchazao' (SCZ) were collected from the tea germplasm plantation of Wuyi University (Wuyishan City, Fujian, China; 27°73′17″ N, 118°00′18″ E) for detection of released volatiles and transcriptome analysis. Indeed, all tea plants were grown under the same cultivation practices. Three independent biological replicates were set up. The collected samples were immediately frozen with liquid nitrogen and stored in a freezer at −80 °C.
Analysis of volatile metabolites
-
The method for determining and analysing volatile metabolites was consistent with our previous report[2]. In brief, the samples were ground into powder in liquid nitrogen, and then 1 g of the powder was immediately transferred to a 20 mL Agilent headspace vial (CA, USA) containing saturated NaCl and 10 µL (50 µg/mL) [2H3]-β-ionone internal standard solution. After 5 min of constant temperature at 100 °C, 120 μm DVB/CAR/PDMS extraction head was inserted into the headspace bottle, and the headspace extraction was carried out for 15 min, and the sample was analyzed at 250 °C for 5 min. The volatile metabolites were detected using an Agilent Model 8890 GC and a 5977B mass spectrometer (Agilent). The analytical conditions were set as follows: desorption of the volatiles from the fibre coating at 250 °C for 5 min in the splitless mode. The carrier gas was helium, and the linear velocity was 1.0 mL/min. The temperature of the injector was kept at 250 °C, and the temperature of the detector was kept at 280 °C. Mass spectra were recorded in electron impact ionization mode at 70 eV. The quadrupole mass detector, ion source, and transfer line temperatures were set at 150, 230, and 280 °C, respectively. Mass spectra were scanned in the range m/z 50−500 amu at 1 s intervals.
Detection of volatile compounds
-
Volatile metabolites were identified by comparing the mass spectra with the data system library (MWGC or NIST) and the linear retention index. Each sample was repeated three times, and the data are expressed as the mean ± standard deviation. The concentrations of volatile compounds in tea plants were quantified based on their peak areas and the peak area of the internal standard compound. The bar charts were made by Excel, and the line charts were made by GraphPad Prism 9.0. Analysis of variance and significant difference analysis were performed by SPSS 26.0. Principal component analysis (PCA) of the identified metabolites was performed using the R package (
www.r-project.org ). Based on the variable importance in project (VIP) score obtained by the OPLS-DA model, metabolites with VIP ≥ 1.0 and fold change (FC) ≥ 1.5 or FC ≤ 0.67 were defined as significantly changed metabolites (SCMs). The calculation method for the odour activity values (OAVs) was the same as that used in a previous study. OAV = C/OT, where C is the concentration of the volatile compound and OT is its odour threshold[22]. Compounds with OAV ≥ 1 were considered potential contributors to the tea aroma profile.Transcriptome-based analysis of aroma component-related pathways and differentially expressed genes
-
The transcriptome data was based on contemporaneous data that we previously published[18]. All RNA-seq data are publicly available in the BIG Data Center (
https://bigd.big.ac.cn ) under project number PRJCA009753. Differential expression analysis with DESeq2 software. Genes with |log2FC| ≥ 1 and p-value < 0.05 were considered to be differentially expressed genes (DEGs) by the DESeq2 R package[23]. All the genes assembled by the transcriptome were compared with six databases (NR, Swiss-Prot, Pfam, EggNOG, GO, and KEGG) to obtain the functional information of genes, and the annotation of each database was statistically analysed. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed using the ClusterProfiler v4.0.0 R package[24] and BLAST was set to e-value ≤ 10−5. For TPS gene, the hidden Markov models of PF01397 and PF03936 were downloaded from Pfam (http://pfam.xfam.org/ ) database, and HMMER software was used to search the TPS gene family sequence. The BLASTP of NCBI and Swiss-Prot was used to predict the possible function of CsTPS gene, and the threshold was set as E-value < 10−5 and identity > 90%. For transcription factors (TFs), all the genes were annotated in the Plant Transcription Factor Database (PlantTFDB v5.0) to determine whether they are TFs. TBtools software was used to make a heatmap for visualization of DEGs.Gene coexpression analysis
-
Weighted gene coexpression network analysis (WGCNA) was performed using the WGCNA R package. Genes with TPM > 1 and coefficient of variation (cv) > 0.1 were used to construct the coexpression network. After filtering, the abundance of 11,222 genes and 20 metabolites was used to build a signed coexpression network by calculating Pearson correlations. The soft-thresholding power of the correlation network was set to 14, the minimum module size was equal to 30, and the minimum height for merging modules was set to 0.5. The module networks were visualized using Gephi software.
Quantitative real-time PCR verification
-
cDNA synthesis and qRT‒PCR tests were performed to verify the reliability of the RNA-Seq data according to previous methods[25]. CsGAPDH (GE651107) was used as a reference control, and the primers of validated genes were designed using Primer3Plus (
www.primer3plus.com ). The primer information is listed in Supplemental Table S1. All samples were analysed in three biological replicates. The relative expression level was calculated using the 2−ΔΔCᴛ method[26]. -
In total, 88 volatiles were identified by GC–MS in the seven tea cultivars (Table 1), including alcohols (22), phenols (3), aldehydes (12), acids (5), terpenoids (13), ketones (7), hydrocarbons (3), heterocyclic compounds (3) and esters (20). Alcohols and esters accounted for 47.72% of the total aroma content, among which geraniol, linalool, methyl salicylate and (E)-3-hexen-1-ol acetate had higher contents. There were 19 volatiles with relative contents greater than 100 µg/kg. Geraniol and linalool contain more than 1,000 µg/kg, accounting for 1%−5% of the total content. The total contents of volatiles in the seven tea cultivars were in the following order: JX > FDDB > SCZ > FJSX > LJ43 > TGY > BHZ.
Table 1. The volatile components of seven tea cultivars.
ID CAS RT /min Compounds Relative contents (µg/kg) TGY JX FJSX FDDB BHZ LJ43 SCZ 1 106-24-1 14.27 Geraniol 1,358.90 ± 49.69d 5103.36 ± 85.9a 3,017.74 ± 80.37b 2,573.91 ± 69.41c 239.72 ± 10.4e 1,321.78 ± 27.49d 2,510.78 ± 62.78c 2 78-70-6 11.46 Linalool 1,012.66 ± 10.81d 1,258.39 ± 74.46bc 1,106.32 ± 71.3cd 2,465.09 ± 167.61a 478.07 ± 44.92e 1,212.96 ± 18.69bcd 1,408.27 ± 132.44b 3 40716-66-3 19.76 (E)-Nerolidol 54.73 ± 2.25c 54.71 ± 0.86c 72.67 ± 5.77b 34.06 ± 1.59d 107.39 ± 4.41a 67.08 ± 3.28b 115.07 ± 5.96a 4 106-25-2 13.75 Nerol 22.42 ± 0.62e 72.25 ± 1.44a 49.45 ± 4.07cd 60.73 ± 1.75b 11.29 ± 0.31f 35.87 ± 0.79d 60.29 ± 3.10b 5 928-96-1 6.67 (Z)-3-Hexenol 34.90 ± 0.64d 33.82 ± 1.24d 79.71 ± 11.02c 177.94 ± 10.84b 73.79 ± 9.13c 170.01 ± 4.20b 229.24 ± 26.97a 6 100-51-6 12.50 Benzyl alcohol 0.28 ± 0.02a 0.16 ± 0.03cd 0.16 ± 0.03bcd 0.22 ± 0.03ab 0.12 ± 0.01d 0.21 ± 0.00bc 0.17 ± 0.02bcd 7 60-12-8 11.69 Phenylethyl alcohol 95.04 ± 6.81e 114.96 ± 1.21e 15.49 ± 0.09f 739.79 ± 17.52a 414.46 ± 7.66c 338.25 ± 5.13d 689.34 ± 36.16b 8 505-32-8 23.78 Isophytol 10.36 ± 2.57b 9.28 ± 1.52b 12.55 ± 2.90b 7.46 ± 2.43b 24.32 ± 8.12a 8.23 ± 3.84b 14.38 ± 3.96ab 9 150-86-7 24.63 Phytol 11.05 ± 3.74ab 9.64 ± 1.83ab 18.00 ± 6.78ab 7.29 ± 3.37b 30.89 ± 18.38a 6.36 ± 4.45b 15.20 ± 9.07ab 10 29171-23-1 24.77 Dehydroisophytol 2.73 ± 0.71b 1.83 ± 0.39b 3.36 ± 1.03ab 1.71 ± 0.63b 5.53 ± 1.83a 1.73 ± 0.93b 2.86 ± 0.96b 11 143-08-8 12.74 1-Nonanol 10.40 ± 0.33c 8.33 ± 0.59cd 6.59 ± 0.56d 29.29 ± 0.77a 21.15 ± 1.31b 31.58 ± 1.08a 31.27 ± 2.04a 12 111-70-6 9.03 1-Heptanol 6.53 ± 0.27d 5.88 ± 0.27d 3.77 ± 0.51d 19.03 ± 1.98a 11.75 ± 1.10bc 10.68 ± 0.49c 13.75 ± 2.12b 13 10482-56-1 13.24 α-Terpineol 26.66 ± 0.24a 26.81 ± 1.26a 19.35 ± 1.44b 24.79 ± 1.22a 6.21 ± 0.53e 10.88 ± 0.71d 14.51 ± 1.87c 14 10340-23-5 12.43 (Z)-3-Nonen-1-ol 0.97 ± 0.13d 1.44 ± 0.12bc 1.22 ± 0.16cd 2.26 ± 0.09a 1.27 ± 0.05cd 2.34 ± 0.17a 1.75 ± 0.21b 15 2425-77-6 22.13 2-Hexyl-1-decanol 2.22 ± 0.05a 1.22 ± 0.11d 1.85 ± 0.35bc 1.63 ± 0.08c 2.05 ± 0.07ab 1.64 ± 0.02c 1.91 ± 0.02bc 16 481-34-5 21.18 α-Cadinol 3.08 ± 0.08d 11.32 ± 0.47a 4.30 ± 0.28c 1.89 ± 0.13e 5.97 ± 0.16b 2.18 ± 0.09e 1.67 ± 0.14e 17 5944-20-7 13.83 Isogeraniol 5.75 ± 0.21e 7.19 ± 0.26e 23.45 ± 2.80b 16.03 ± 1.19c 11.98 ± 0.87d 30.30 ± 0.99a 14.74 ± 1.49cd 18 498-00-0 14.09 Vanillyl alcohol 246.47 ± 2.27b 242.40 ± 4.17b 251.82 ± 8.47ab 257.91 ± 7.73ab 243.94 ± 6.00b 265.31 ± 4.52a 244.78 ± 11.91b 19 15051-81-7 19.43 epi-g-Eudesmol 1.09 ± 0.10d 0.90 ± 0.09de 1.13 ± 0.10cd 0.66 ± 0.06e 1.76 ± 0.14ab 1.43 ± 0.13bc 1.79 ± 0.29a 20 18675-33-7 15.29 (+)-Neodihydrocarveol 2.54 ± 0.12a 1.61 ± 0.06b 0.79 ± 0.04d 0.49 ± 0.02e 1.02 ± 0.11c 0.00 ± 0.00f 0.00 ± 0.00f 21 5989-33-3 10.90 (Z)-Linalool oxide (furanoid) 26.59 ± 1.08e 97.88 ± 5.72d 43.45 ± 4.25e 255.98 ± 13.68a 84.79 ± 6.13d 202.69 ± 13.96b 130.23 ± 11.66c 22 34995-77-2 11.20 (E)-Linalool oxide (furanoid) 81.38 ± 4.35e 314.5 ± 19.75c 81.20 ± 5.77e 666.69 ± 29.84a 189.5 ± 12.55d 408.19 ± 31.09b 299.17 ± 27.17c 23 39028-58-5 12.84 (E)-Linalool oxide (pyranoid) 15.78 ± 0.36d 40.13 ± 2.58b 12.91 ± 0.95d 69.64 ± 0.99a 39.31 ± 2.55b 34.67 ± 1.30c 36.72 ± 1.82bc Alcohols (23) 3,032.53 ± 74.37e 7,418.01 ± 149.51a 4,827.26 ± 203.54c 7,414.48 ± 291.74a 2,006.27 ± 48.02f 4,164.36 ± 50.25d 5,837.9 ± 253.16b 24 97-53-0 16.12 Eugenol 3.17 ± 0.58a 0.73 ± 0.01c 1.82 ± 0.44b 1.67 ± 0.13b 0.95 ± 0.05c 1.94 ± 0.04b 2.01 ± 0.14b 25 3228-02-2 15.13 3-Methyl-4-isopropylphenol 5.46 ± 1.38a 1.03 ± 0.11c 3.14 ± 0.68b 1.82 ± 0.07bc 0.76 ± 0.12c 0.99 ± 0.15c 0.87 ± 0.18c Phenols (2) 8.63 ± 1.85a 1.76 ± 0.12c 4.96 ± 1.11b 3.49 ± 0.09bc 1.71 ± 0.14c 2.93 ± 0.14c 2.88 ± 0.29c 26 141-27-5 14.55 Neral 25.34 ± 1.36c 93.8 ± 3.25a 60.48 ± 7.92b 65.70 ± 7.92b 5.30 ± 0.95d 37.78 ± 1.06c 59.69 ± 6.84b 27 121-33-5 13.69 Vanillin 3.25 ± 0.28d 0.74 ± 0.11e 10.16 ± 2.30a 5.28 ± 0.45bcd 3.90 ± 0.21cd 6.27 ± 0.86b 6.15 ± 0.11bc 28 34246-54-3 12.67 3-Ethylbenzaldehyde 0.65 ± 0.01cd 0.62 ± 0.02de 0.42 ± 0.05e 0.70 ± 0.07cd 0.86 ± 0.12bc 1.01 ± 0.02ab 1.10 ± 0.11a 29 6728-26-3 14.46 (E)-2-Hexenal 3.25 ± 0.48cd 5.99 ± 0.58a 4.38 ± 0.19b 4.10 ± 0.31bc 1.93 ± 0.12e 3.16 ± 0.37d 4.01 ± 0.36bcd 30 100-52-7 8.87 Benzaldehyde 9.82 ± 0.70ab 8.20 ± 0.37bc 6.17 ± 1.08c 8.72 ± 1.21bc 9.00 ± 1.21bc 11.07 ± 0.73ab 12.86 ± 2.30a 31 122-78-1 11.69 Phenylacetaldehyde 99.41 ± 7.28e 121.13 ± 1.40e 18.87 ± 0.40f 746.69 ± 12.32a 435.00 ± 6.55c 356.40 ± 14.09d 700.08 ± 30.99b 32 112-31-2 13.40 Decanal 0.59 ± 0.07b 0.64 ± 0.15b 0.69 ± 0.07b 0.82 ± 0.17b 0.86 ± 0.14b 1.19 ± 0.12a 0.81 ± 0.04b 33 111-71-7 7.67 Heptanal 143.12 ± 4.70a 2.18 ± 0.24b 2.16 ± 0.41b 1.60 ± 0.24b 1.15 ± 0.19b 1.09 ± 0.11b 0.90 ± 0.11b 34 66-25-1 5.45 Hexanal 7.83 ± 1.19d 15.44 ± 3.43bcd 12.48 ± 1.33cd 18.63 ± 2.71bc 27.58 ± 6.03a 29.47 ± 3.54a 22.46 ± 1.58ab 35 123-72-8 5.45 Butanal 6.59 ± 1.30d 14.07 ± 3.66bcd 11.59 ± 1.53cd 16.53 ± 1.67bc 26.64 ± 5.32a 28.38 ± 3.38a 21.63 ± 2.66ab 36 110-62-3 4.83 Valeraldehyde 14.41 ± 1.13de 20.84 ± 1.88cd 13.41 ± 2.76e 24.21 ± 1.66c 33.52 ± 2.5ab 31.13 ± 1.22b 40.15 ± 5.75a 37 590-86-3 5.03 Isovaleraldehyde 48.88 ± 1.45a 21.11 ± 1.69b 33.19 ± 5.46ab 37.00 ± 5.6ab 34.38 ± 8.89ab 45.65 ± 1.09a 39.15 ± 11.95a Aldehydes (12) 363.15 ± 9.42c 304.75 ± 8.93c 174.01 ± 3.96d 929.96 ± 12.47a 580.11 ± 28.65b 552.59 ± 8.81b 908.99 ± 56.08a 38 459-80-3 16.02 Geranic acid 37.94 ± 9.28bc 115.49 ± 33.01a 62.74 ± 23.82b 20.81 ± 3.51bc 8.82 ± 2.85c 19.69 ± 2.49c 131.88 ± 17.95a 39 112-05-0 14.48 Nonanoic acid 2.58 ± 0.66c 4.45 ± 1.22ab 4.80 ± 0.65ab 4.39 ± 0.67ab 5.51 ± 0.47a 3.51 ± 0.44bc 4.64 ± 0.40ab 40 111-14-8 12.69 Heptanoic acid 119.89 ± 0.98a 101.69 ± 5.30cd 115.81 ± 5.88ab 107.27 ± 3.55bcd 98.31 ± 5.48d 111.4 ± 2.49abc 104.84 ± 6.82bcd 41 109-52-4 14.48 Pentanoic acid 4.92 ± 0.63b 7.14 ± 1.45ab 8.07 ± 1.02a 8.00 ± 1.74a 8.12 ± 1.10a 6.55 ± 0.54ab 7.57 ± 0.45ab 42 79-09-4 12.05 Propanoic acid 65.13 ± 26.72a 79.82 ± 37.7a 89.19 ± 28.57a 50.37 ± 21.63a 65.56 ± 23.49a 64.99 ± 18.33a 67.71 ± 24.17a Acids (5) 230.47 ± 31.08ab 308.59 ± 71.53a 280.6 ± 47.71ab 190.84 ± 20.11b 186.32 ± 20.63b 206.13 ± 16.59b 316.64 ± 32.43a 43 127-41-3 9.92 α-Ionone 12.05 ± 0.88cd 23.28 ± 0.54a 14.7 ± 1.06bc 17.81 ± 1.66b 2.60 ± 0.43e 10.34 ± 1.64d 14.11 ± 2.26c 44 79-77-6 18.39 β-Ionone 87.2 ± 1.64c 90.97 ± 0.81c 92.93 ± 7.34c 103.48 ± 5.56b 111.25 ± 4.28ab 108.43 ± 2.05ab 114.18 ± 2.52a 45 689-67-8 17.79 Geranylacetone 10.67 ± 0.30bc 12.01 ± 0.49a 11.15 ± 1.07abc 10.07 ± 0.57c 11.79 ± 0.19ab 10.58 ± 0.05bc 11.43 ± 0.29ab 46 502-69-2 23.10 Fitone 6.01 ± 0.21bc 4.82 ± 0.37de 6.70 ± 0.61b 7.05 ± 0.24b 9.15 ± 0.70a 4.17 ± 0.21e 5.51 ± 0.61cd 47 471-15-8 11.62 3-Thujone 11.55 ± 3.73a 1.49 ± 1.68b 6.56 ± 2.92ab 0.84 ± 0.34b 4.92 ± 4.11b 2.18 ± 1.52b 0.65 ± 0.44b 48 488-10-8 16.84 Jasmone 6.64 ± 0.35d 4.79 ± 0.61e 4.24 ± 0.28e 5.18 ± 0.26e 9.53 ± 0.41b 7.84 ± 0.53c 12.43 ± 0.39a 49 23726-93-4 16.53 β-Damascenone 1.01 ± 0.15e 18.05 ± 0.75a 14.26 ± 1.60b 7.58 ± 0.13cd 1.82 ± 0.24e 6.11 ± 0.14d 9.02 ± 0.31c Ketones (7) 135.12 ± 4.62c 155.41 ± 3.29ab 150.53 ± 12.41bc 152.02 ± 6.26ab 151.07 ± 7.13bc 149.65 ± 0.72bc 167.33 ± 4.26a 50 544-76-3 20.41 Hexadecane 3.25 ± 0.34a 4.04 ± 0.38a 3.41 ± 0.90a 4.73 ± 1.33a 4.82 ± 0.68a 4.11 ± 0.54a 4.26 ± 0.56a 51 629-59-4 16.97 Tetradecane 7.30 ± 00.43ab 5.87 ± 0.54b 6.21 ± 0.46b 7.82 ± 1.40ab 7.82 ± 0.95ab 8.41 ± 0.70a 7.27 ± 0.84ab 52 3891-99-4 18.03 2,6,10-Trimethyltridecane 6.51 ± 0.12bc 5.13 ± 0.72c 6.91 ± 2.37abc 6.36 ± 0.50bc 8.50 ± 0.66ab 8.21 ± 0.16ab 9.67 ± 1.60a Hydrocarbons (3) 17.07 ± 0.59ab 15.04 ± 1.09b 16.54 ± 3.61ab 18.9 ± 2.56ab 21.14 ± 2.03a 20.72 ± 1.21ab 21.2 ± 2.99a 53 120-72-9 15.05 Indole 0.00 ± 0.00f 36.36 ± 1.23b 0.00 ± 0.00f 11.8 ± 0.78d 42.41 ± 1.36a 7.78 ± 0.62e 24.75 ± 2.46c 54 91-64-5 17.72 Coumarin 16.81 ± 1.45a 18.38 ± 0.34a 17.67 ± 1.54a 11.5 ± 0.53b 10.28 ± 0.58b 11.43 ± 0.48b 10.50 ± 0.61b 55 36431-72-8 15.22 Theaspirane 3.92 ± 0.05c 10.98 ± 0.92b 4.57 ± 0.49c 14.31 ± 1.95a 5.88 ± 0.24c 9.10 ± 0.19b 4.23 ± 0.44c Heterocyclic compounds (3) 20.73 ± 1.41f 65.72 ± 0.98a 22.24 ± 1.79f 37.61 ± 2.32d 58.57 ± 0.92b 28.31 ± 0.70e 39.48 ± 2.71c 56 6753-98-6 18.06 α-Humulene 6.58 ± 0.40a 4.63 ± 0.11b 1.16 ± 0.04e 1.19 ± 0.07e 3.06 ± 0.21d 0.86 ± 0.04e 3.68 ± 0.42c 57 87-44-5 17.43 β-Caryophyllene 14.30 ± 0.46a 14.97 ± 0.30a 2.04 ± 0.28d 4.54 ± 0.73c 8.49 ± 0.48b 0.50 ± 0.19e 1.62 ± 0.26d 58 502-61-4 18.79 α-Farnesene 28.28 ± 0.62c 34.79 ± 0.54b 12.15 ± 1.56e 14.98 ± 1.35e 43.86 ± 1.15a 12.50 ± 0.69e 24.02 ± 1.91d 59 123-35-3 9.39 Myrcene 241.44 ± 15.17c 669.11 ± 7.54a 374.82 ± 41.18b 426.14 ± 44.52b 44.16 ± 6.77d 230.59 ± 33.5c 363.94 ± 50.74b 60 3779-61-1 10.44 (E)-β-Ocimene 107.36 ± 5.92c 263.91 ± 3.58a 147.98 ± 15.26b 174.1 ± 17.53b 23.92 ± 4.42d 99.25 ± 13.74c 155.4 ± 25.28b 61 7216-56-0 11.95 Allo-ocimene 14.16 ± 1.00c 37.51 ± 0.65a 21.17 ± 2.50b 23.85 ± 2.91b 2.43 ± 0.34d 12.92 ± 2.22c 20.17 ± 2.98b 62 99-85-4 10.68 γ-Terpinene 11.22 ± 1.12b 17.20 ± 0.51a 12.16 ± 0.68b 13.79 ± 1.33b 2.42 ± 0.36d 8.25 ± 1.30c 11.29 ± 2.04b 63 5208-59-3 16.78 β-Bourbonene 4.85 ± 0.11d 4.44 ± 0.06d 12.08 ± 2.07b 8.51 ± 1.21c 17.81 ± 1.93a 5.83 ± 0.16cd 3.54 ± 0.47d 64 29050-33-7 9.91 4-Carene 12.04 ± 0.92cd 23.33 ± 0.51a 14.74 ± 1.06bc 17.81 ± 1.66b 2.66 ± 0.40e 10.34 ± 1.64d 14.13 ± 2.31c 65 483-76-1 19.10 δ-Cadinene 12.99 ± 0.13c 93.21 ± 2.13a 7.78 ± 0.97de 6.12 ± 0.41e 9.49 ± 0.47d 17.99 ± 0.70b 6.78 ± 0.55e 66 21391-99-1 19.51 α-Calacorene 10.27 ± 0.10b 42.73 ± 0.64a 6.58 ± 0.53d 4.65 ± 0.21e 8.12 ± 0.70c 10.1 ± 0.44b 5.53 ± 0.59de 67 483-77-2 19.16 Calamenene 6.57 ± 0.15c 61.3 ± 1.03a 4.16 ± 0.49de 4.66 ± 0.12de 5.46 ± 0.70cd 16.02 ± 0.98b 3.38 ± 0.62e 68 5989-27-5 12.19 d-Limonene 5.38 ± 0.38cd 14.19 ± 0.42a 8.26 ± 0.99b 8.95 ± 1.46b 0.94 ± 0.23e 4.99 ± 0.79d 7.49 ± 1.14bc Terpenoids (13) 475.44 ± 24.58c 1281.32 ± 15.03a 625.07 ± 57.13b 709.29 ± 67.59b 172.83 ± 17.3d 430.14 ± 56.04c 620.98 ± 89.21b 69 3681-82-1 9.64 (E)-3-Hexen-1-ol acetate 125.08 ± 1.45d 223.82 ± 15.90cd 1030.93 ± 119a 690.15 ± 92.67b 398.2 ± 79.2c 981.62 ± 42.12a 871.21 ± 172.5ab 70 61444-38-0 16.64 (Z)-3-Hexenyl (Z)-3-hexenoate 10.90 ± 0.08d 10.16 ± 0.50d 46.93 ± 2.48b 48.58 ± 3.24b 7.03 ± 0.63d 23.17 ± 1.03c 54.08 ± 3.55a 71 31501-11-8 16.57 (E)-hex-3-enyl hexanoate 5.27 ± 0.10e 23.15 ± 0.86d 76.15 ± 6.66a 26.00 ± 2.08d 22.04 ± 1.86d 43.61 ± 1.26b 36.01 ± 2.91c 72 2497-18-9 9.82 (E)-2-Hexenyl acetate 0.00 ± 0.00c 1.48 ± 0.15bc 8.98 ± 1.42a 2.34 ± 0.40bc 7.07 ± 1.83a 0.00 ± 0.00c 3.23 ± 0.91b 73 65405-77-8 22.84 (Z)-3-Hexenyl salicylate 0.12 ± 0.01d 4.60 ± 0.07a 4.55 ± 0.53a 2.40 ± 0.32c 4.28 ± 0.37b 3.78 ± 0.16b 2.22 ± 0.21d 74 41519-23-7 12.99 (Z)-3-Hexenyl isobutyrate 9.20 ± 0.41d 20.40 ± 1.44c 83.98 ± 8.54a 12.03 ± 1.63cd 12.81 ± 1.82cd 39.75 ± 1.61b 34.51 ± 4.53b 75 53398-85-9 14.82 (Z)-3-Hexenyl 2-methylbutyrate 0.00 ± 0.00e 0.22 ± 0.02d 1.23 ± 0.15a 0.21 ± 0.02d 0.28 ± 0.02d 0.87 ± 0.06b 0.53 ± 0.08c 76 35852-46-1 13.90 (Z)-3-Hexenyl valerate 0.23 ± 0.05e 0.61 ± 0.10e 5.30 ± 0.89a 1.89 ± 0.32cd 3.32 ± 0.51b 2.15 ± 0.05c 1.05 ± 0.13de 77 1189-09-9 15.51 Methyl geranate 3.73 ± 0.18de 28.76 ± 1.08a 23.44 ± 2.68b 5.40 ± 0.07d 3.02 ± 0.09de 1.48 ± 0.24e 10.69 ± 0.77c 78 150-84-5 23.07 Citronellyl acetate 26.23 ± 4.83bc 22.86 ± 3.66bc 24.37 ± 1.79bc 15.38 ± 4.07c 50.53 ± 12.99a 18.84 ± 7.95bc 32.85 ± 6.84b 79 2051-49-2 16.72 Hexyl hexanoate 0.00 ± 0.00e 2.77 ± 0.15c 9.44 ± 1.23a 1.56 ± 0.19cd 4.97 ± 0.39b 0.97 ± 0.05de 1.18 ± 0.08de 80 142-92-7 9.78 Hexyl acetate 0.00 ± 0.00d 1.57 ± 0.27cd 8.85 ± 1.48a 2.79 ± 0.49bc 4.39 ± 1.06b 2.59 ± 0.17bc 1.92 ± 0.54c 81 120-51-4 22.56 Benzyl benzoate 2.17 ± 0.12cd 4.02 ± 0.53a 2.95 ± 0.53bc 3.00 ± 0.45bc 3.85 ± 0.42b 1.96 ± 0.18d 2.52 ± 0.27cd 82 110-27-0 22.96 Isopropyl myristate 0.06 ± 0.01e 2.55 ± 0.18a 1.35 ± 0.12bc 1.34 ± 0.07bc 1.14 ± 0.07cd 1.03 ± 0.03d 1.44 ± 0.06b 83 606-45-1 15.77 Methyl 2-methoxybenzoate 7.30 ± 0.20a 0.56 ± 0.03e 3.00 ± 0.26b 2.02 ± 0.08c 0.42 ± 0.03e 1.01 ± 0.02d 0.97 ± 0.11d 84 102-16-9 14.94 Benzyl phenylacetate 0.97 ± 0.07b 1.64 ± 0.04a 1.07 ± 0.19b 0.73 ± 0.07c 0.38 ± 0.02d 0.49 ± 0.02d 0.74 ± 0.05c 85 7011-83-8 16.72 Dihydrojasmone lactone 0.00 ± 0.00e 3.14 ± 0.11c 11.60 ± 1.36a 1.65 ± 0.18d 5.14 ± 0.36b 1.36 ± 0.02d 1.52 ± 0.11d 86 25524-95-2 16.57 Jasmine lactone 5.34 ± 0.11e 23.23 ± 0.92d 76.66 ± 6.57a 26.21 ± 1.94d 22.16 ± 1.84d 43.74 ± 1.33b 36.1 ± 2.91c 87 1211-29-6 23.24 Methyl jasmonate 12.81 ± 2.06b 13.26 ± 2.07b 12.50 ± 1.11b 8.73 ± 1.89b 27.01 ± 4.9a 10.85 ± 4.47b 15.19 ± 1.67b 88 119-36-8 13.19 Methyl salicylate 294.81 ± 22.12d 2,085.7 ± 131.83a 307.14 ± 36.23d 498.63 ± 15.53c 764.22 ± 42.52b 516.16 ± 30.26c 483.65 ± 46.37c Esters (20) 504.25 ± 27.11d 2,474.51 ± 157.38a 1,740.43 ± 183.45b 1,351.05 ± 114.32c 1,342.27 ± 103.23c 1,695.42 ± 61.37b 1,591.61 ± 204.25bc All data are shown as the mean ± standard deviation (SD). Significant differences among various groups are represented by different letters (p < 0.05).. RT: retention time. The phenotypes of seven tea cultivars are shown in Fig. 1a. The PCA score plot of the main chemical components indicated that the first two principal components explained 27.5% and 21.3% of the total variance, respectively, and the cumulative variance contribution reached 49.8% (Fig. 1b), which indicated that PC1 and PC2 were selected to analyze the samples with good reliability. The seven tea cultivars showed different distribution characteristics in the PCA score plot: JX, BHZ, and TGY were far away from other cultivars; FDDB and FJSX were relatively close; and LJ43 and SCZ were relatively close.
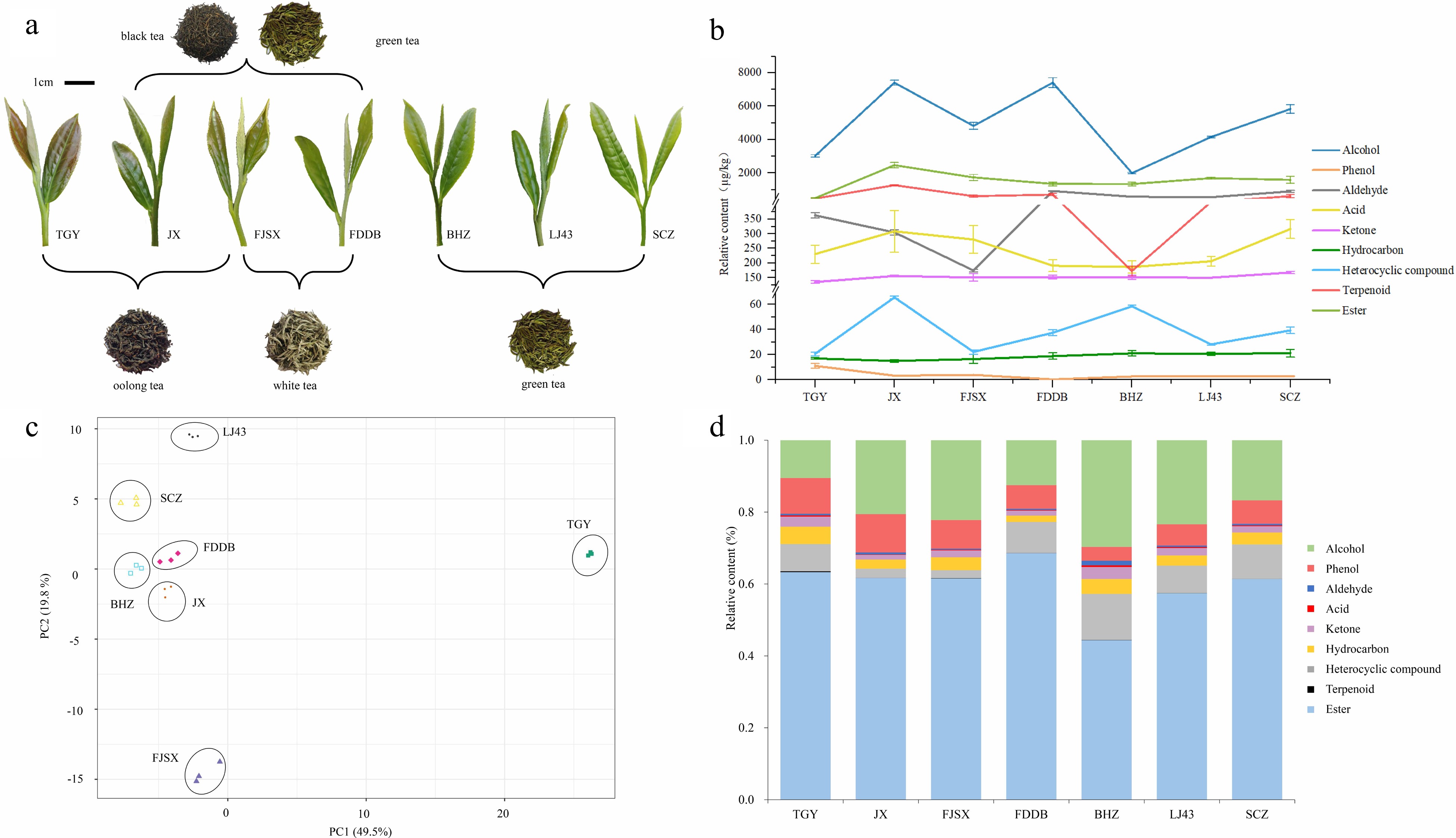
Figure 1.
Multivariate statistical analysis of volatile components of seven tea cultivars. (a) Phenotypes of one bud and two leaves and their suitability. (b) PCA principal component analysis. (c) Types and relative contents of volatile components. (d) Proportions of volatile components.
The comparative analysis identified differences in the relative content of volatiles in the seven tea cultivars (Fig. 1c, d). The volatile aroma substances with the highest contents in TGY were mainly phenols. In particular, eugenol had the highest levels of contents in TGY as compared to other cultivars. Among these compounds, alcohols and esters were present in the greatest numbers, indicating major contributions to aroma. The content of alcohols and esters in JX were higher than in other cultivar aroma types, nerol, geraniol, myrcene, (E)-β-ocimene, δ-cadinene and d-limonene had higher concentrations in JX than other cultivars. Compared with other cultivars, these aroma categories varied in FJSX was not abundant. Interestingly, Jasmine lactone had the highest concentrations in FJSX. The representative green tea cultivars showed higher contents of alcohol and aldehyde. In the alcohol group, linalool had the highest concentrations in FDDB than other cultivars, (E)-nerolidol and (Z)-3-hexenol had the highest concentrations in SCZ than other cultivars. The content of aldehydes in FDDB and SCZ than other cultivars, for example, phenylacetaldehyde had higher concentrations than other cultivars. In the aldehyde group, hexanal and butanal had the highest concentrations in LJ43. It is worth noting that indole had the highest concentrations in BHZ than other cultivars.
Analysis of differential volatile components of seven tea cultivars
-
A total of 54 significantly changed metabolites (SCMs) were identified in seven tea cultivars (Fig. 2). Specifically, the study found that the proportion of up-regulated terpenoids was highest in JX, while the proportion of down-regulated terpenoids was highest in BHZ. Among the esters, the proportion of up-regulated compounds was highest in FJSX, while the proportion of down-regulated compounds was highest in TGY. Additionally, the content of three phenolic compounds was significantly up-regulated in TGY, and the content of alcohol compounds was significantly up-regulated in FDDB.
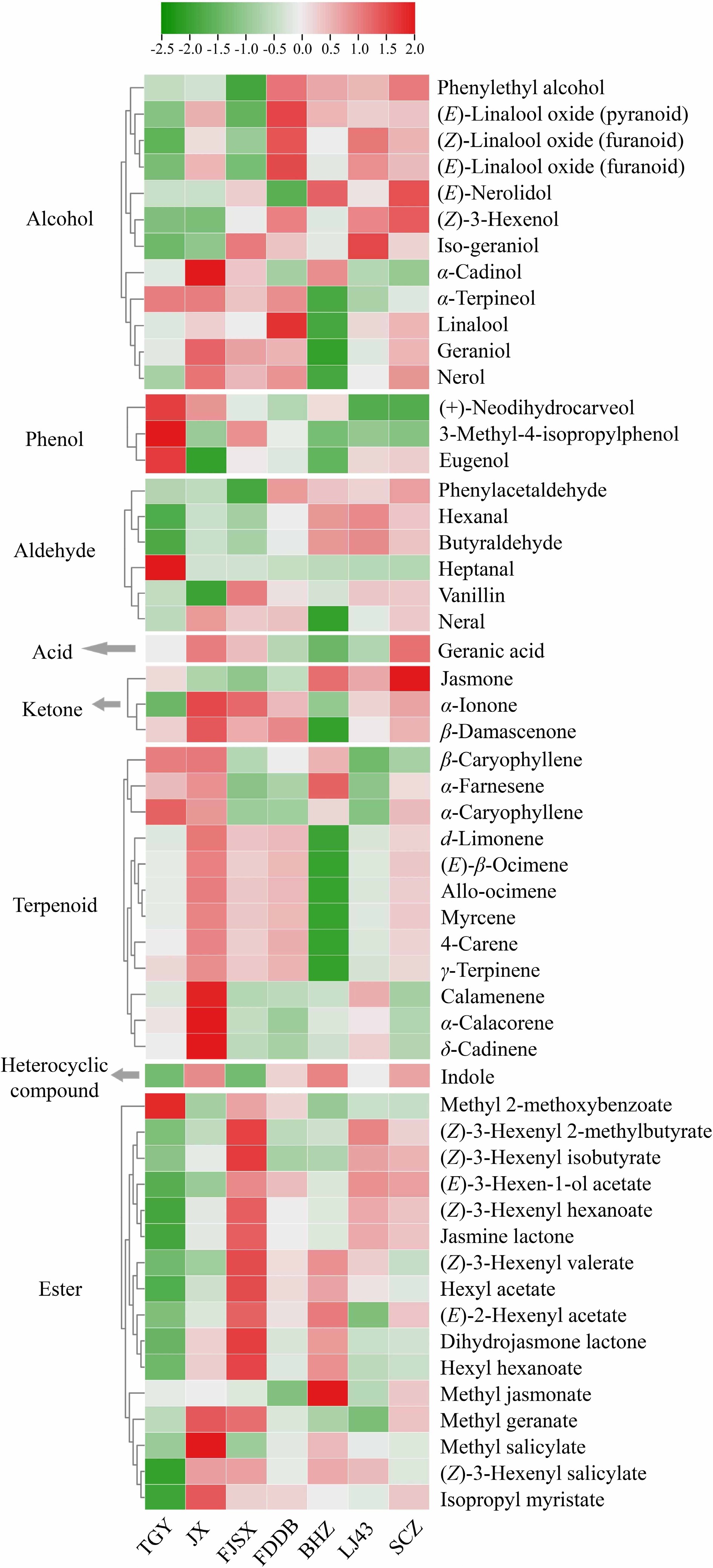
Figure 2.
Analysis of differential volatile components of seven tea cultivars.
Furthermore, the study conducted a more detailed analysis of the compounds with significantly increased content in each tea variety. In TGY and JX, the content of β-caryophyllene and α-caryophyllene was significantly higher than in other cultivars. The highest proportion of eight ester compounds was found in FJSX. Phenylethyl alcohol and phenylacetaldehyde levels showed significant variation in seven cultivars, with the highest levels found in FDDB. In SCZ, LJ43, and BHZ, the content of (Z)-3-hexenol was significantly up-regulated. In SCZ and BHZ, the levels of (E)-nerolidol and jasmone were significantly up-regulated. Finally, the content of indole was significantly accumulated in JX and BHZ, while the levels of α-farnesene and methyl jasmonate were significantly higher in BHZ than in other cultivars.
OAV value of differential volatile components in seven tea cultivars
-
The odor activity values (OAVs) of the identified volatiles are shown in Table 2. A total of 26 volatiles were determined to have OAV > 1 in tea samples, of which two volatiles had OAVs ≥ 1000. The OAVs of β-damascenone (OAV: 503.45-9026.77), β-ionone (OAV: 1245.71-1631.16), geraniol (OAV: 31.96-680.45), linalool (OAV: 79.68-410.85) and phenylacetaldehyde (OAV: 4.74-186.67) were higher than those of other compounds, indicating that they played significant roles in the aroma of the seven tea cultivars.
Table 2. OAV values of differential volatile components in seven tea cultivars.
Volatile
componentsAroma
characteristicsAroma thresholds[3,27−30]
(μg/kg)Relative OAV value TGY JX FJSX FDDB BHZ LJ43 SCZ Geraniol Rosy, sweet 7.5 181.19 680.45 402.37 343.19 31.96 176.24 334.77 Linalool Floral, fruity 6 168.78 209.73 184.39 410.85 79.68 202.16 234.71 (E)-Nerolidol Floral, citrus 15 3.65 3.65 4.84 2.27 7.16 4.47 7.67 Nerol Rosy, orange 49 0.46 1.47 1.01 1.24 0.23 0.73 1.23 (Z)-3-Hexenol Fresh, grassy 110 0.32 0.31 0.72 1.62 0.67 1.55 2.08 Phenylethyl alcohol Floral, rosy 45 2.11 2.55 0.34 16.44 9.21 7.52 15.32 (Z)-Linalool oxide (furanoid) Sweet, floral 190 0.14 0.52 0.23 1.35 0.45 1.07 0.69 (E)-Linalool oxide (furanoid) Sweet, floral 190 0.43 1.66 0.43 3.51 1.00 2.15 1.57 Neral Sweet, fruity 53 0.48 1.77 1.14 1.24 0.10 0.71 1.13 Decanal Sweet, citrus 0.1 5.90 6.38 6.93 8.20 8.60 11.89 8.13 Heptanal Fatty, citrus 10 14.31 0.22 0.22 0.16 0.12 0.11 0.09 Benzaldehyde Almond, nutty 3 3.27 2.73 2.06 2.91 3.00 3.69 4.29 Phenylacetaldehyde Woody, sweet 4 24.85 30.28 4.72 186.67 108.75 89.10 175.02 Hexanal Fresh, fruity, fatty 4.5 1.74 3.43 2.77 4.14 6.13 6.55 4.99 Valeraldehyde Almond, malty 12 1.20 1.74 1.12 2.02 2.79 2.59 3.35 Isovaleraldehyde Fruity 4 12.22 5.28 8.30 9.25 8.59 11.41 9.79 α-Ionone Violet, woody 0.4 30.13 58.20 36.75 44.53 6.50 25.85 35.27 β-Ionone Violet, floral 0.07 1,245.71 1,299.60 1,327.56 1,478.32 1,589.21 1,549.00 1,631.16 Jasmone Jasmine 7 0.95 0.68 0.61 0.74 1.36 1.12 1.78 β-Damascenone Rosy 0.002 503.45 9,026.77 7,127.53 3,791.88 911.92 3,053.12 4,509.23 Indole Floral 40 – 0.91 – 0.29 1.06 0.19 0.62 (E)-β-Ocimene Floral, grassy 34 3.16 7.76 4.35 5.12 0.70 2.92 4.57 Myrcene Fruity, balsamic 15 16.10 44.61 24.99 28.41 2.94 15.37 24.26 (E)-3-Hexen-1-ol acetate Fruity, grassy 31 4.03 7.22 33.26 22.26 12.85 31.67 28.10 Methyl salicylate Herbal, minty 40 7.37 52.14 7.68 12.47 19.11 12.90 12.09 Methyl jasmonate Jasmine 3 4.27 4.42 4.17 2.91 9.00 3.62 5.06 '–' indicates that the OAV value cannot be calculated. There were 21 volatiles with 1 ≤ OAV ≤ 100 in tea samples, of which eight volatiles (phenylethyl alcohol, heptanal, decanal, isovaleraldehyde, α-ionone, myrcene, (E)-3-hexen-1-ol acetate, methyl salicylate) had OAVs ≥ 10. Importantly, we found that heptanal was abundant in TGY with OAV > 1. In total, 13 volatile compounds had OAVs > 1 in tea samples, of which (Z)-3-hexenol (SCZ, FDDB, LJ43), (Z)-linalool oxide (FDDB, LJ43), and jasmone (SCZ, BHZ, LJ43) had OAVs ≥ 1. In addition, indole was determined to have OAVs > 1 in BHZ.
Differentially expressed genes (DEGs) related to three volatile synthesis pathways
-
Terpenoids, unsaturated aliphatic compounds and aromatic compounds are the main aroma components of tea. To explain the mechanism of up- or downregulation of SCMs in seven tea cultivars at the molecular level, we identified DEGs in relevant biosynthetic pathways.
For the terpenoid volatiles synthesis pathway, a total of 20 DEGs and 40 TPS genes were involved in Mevalonate pathway (MVA) and 2-methyl-D-erythritol-4-phosphate pathway (MEP), and six DEGs were involved in the carotenoid metabolic pathway (Fig. 3). Among them, HMGR (CsTGY05G0000313) and HMGS (CsTGY01G0002862) were significantly upregulated 5.55- and 5.46-fold in JX and SCZ, respectively. CsHMGR (3-hydroxy-3-methylglutaryl coenzyme A reductase) and HMGS (3-hydroxy-3-methylglutaryl-CoA synthase) are key rate-limiting enzymes in the MVA of the terpene derivative pathway[31]. LIS/NES (CsTGY08G0001704) and NES/GIS (CsTGY08G0001826) were significantly upregulated 3.09- and 4.7-fold in FDDB and BHZ, respectively. LIS/NES (linalool/nerolidol synthase) and NES/GIS (nerolidol/geranyl linalool synthase) are the key enzyme in the biosynthesis of linalool and nerolidol. (E)-nerolidol has clean and floral aromas, linalool has the fragrance of rose and fruit, and its oxidized products have woody, floral, and camphor odours, which are the main aroma components of tea[3].
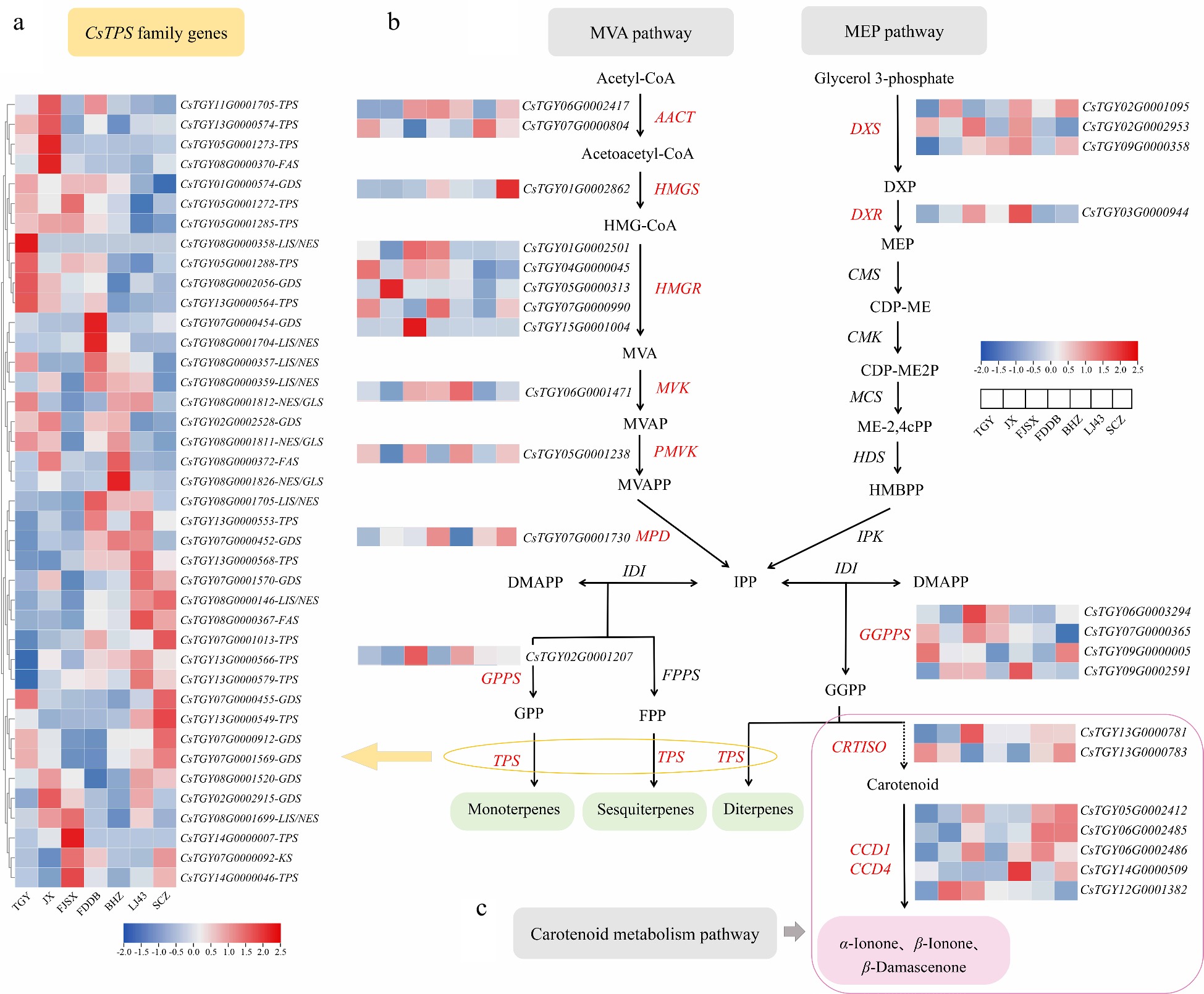
Figure 3.
Expression profiles of genes related to the terpene synthesis pathway. (a) Heatmap of the expression pattern of CsTPS genes. (b) Biosynthetic pathway of terpenes and expression patterns of related DEGs. (c) Metabolic pathway of carotenoids and expression patterns of related DEGs.
For the α-linolenic acid metabolism pathway, 29 DEGs were involved in the α-linolenic acid metabolism pathway (Fig. 4), mainly including acyl-CoA oxidase (ACX), OPC-8:0 CoA ligase (OPCL), allene oxide cyclase (AOC), lipoxygenase (LOX), and alcohol dehydrogenase (ADH). Among these genes, ACX (CsTGY04G0002749) was significantly upregulated 3.76-fold in FDDB. OPCL (CsTGY07G0001858) was only expressed in SCZ. AOC (CsTGY01G0003195) was significantly downregulated 2.35-fold in FJSX, and ADH (CsTGY09G0001886) showed higher expression in TGY and FJSX than in the other tea cultivars. ADH (alcohol dehydrogenase) is a key enzyme responsible for the biosynthesis of the key volatile C6-compounds in green tea leaves, which are important precursors of tea aroma[32]. In particular, (Z)-3-hexenol has grassy odor, (E)-3-hexen-1-ol acetate has grassy and fruity aroma, among which the former is considered to be the main source of green tea aroma[33].
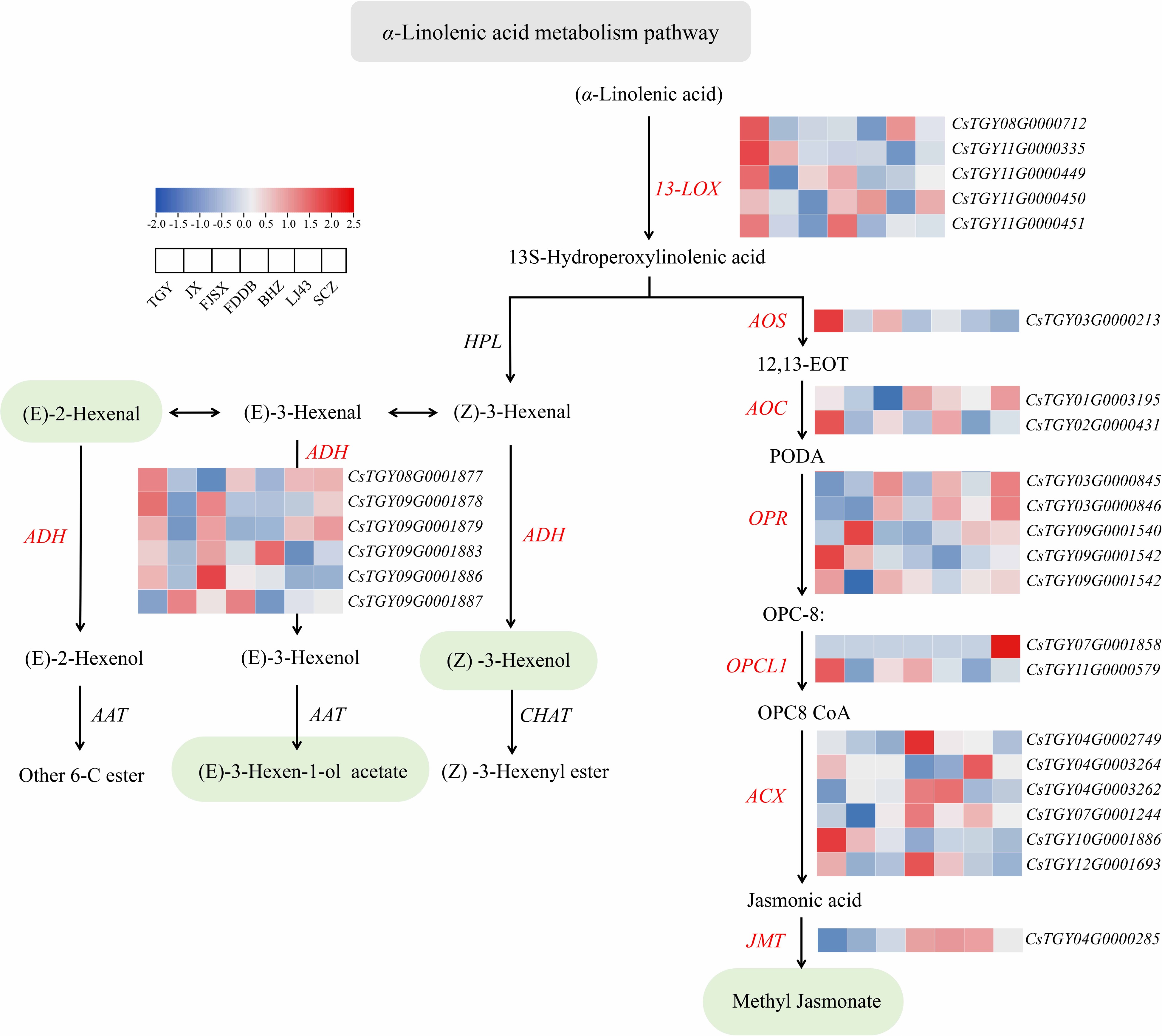
Figure 4.
Expression profiles of genes related to the α-linolenic acid metabolism pathway.
To deeply investigate the mechanisms that regulate the biosynthesis of phenylpropanoid/benzenoids in tea plants, we thoroughly studied the DEGs in the pathways (Fig. 5). In total, 11 DEGs were involved in the phenylpropanoid/benzenoid synthesis pathway, mainly including catechol-O-methyltransferase (COMT), caffeoyl-coenzyme A O-methyltransferase (CCoCOMT), alcohol dehydrogenase (CAD), eugenol synthase (EGS), and benzoic acid carboxyl methyltransferases (BAMT). Among these DEGs, COMT (CsTGY08G0002131), CCoCOMT (CsTGY06G0000958), and CAD (CsTGY04G0002121) showed the highest expression in TGY and were significantly upregulated 3.77-, 7.75-, and 4.63-fold, respectively. COMT, CCoCOMT, and CAD are involved in the biosynthesis of eugenol, which is the main aromatic component of cloves and orchids, with clove aroma[34]. In addition, EGS (CsTGY01G0002412) was significantly upregulated 3.93-fold in JX; BAMT (CsTGY03G0002449, CsTGY03G0002452) was significantly upregulated 3.99- and 52.52-fold in BHZ.
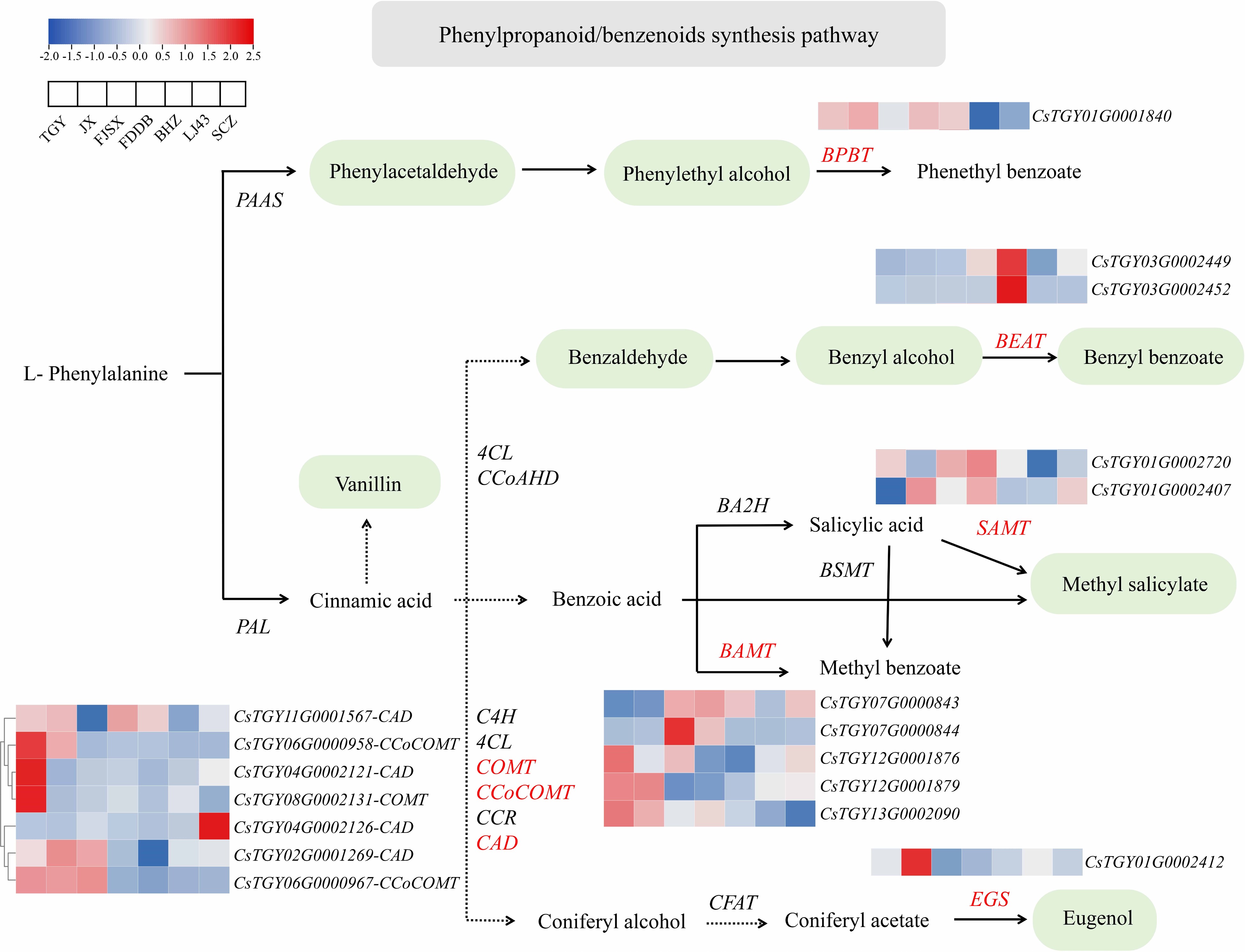
Figure 5.
Expression profiles of genes related to the phenylpropanoid/benzenoid synthesis pathway.
Coexpression network related to key aroma compound formation
-
To understand the gene regulation mechanism of aroma biosynthesis, we correlated 11,222 genes and 20 SCMs were used for WGCNA. After merging similar modules, 14 modules were generated, which comprised 95 to 2,503 genes. Figure 6 showed the correlation between 14 modules and 20 characteristic volatiles. Modules with larger correlation coefficients and smaller p-values are highly correlated phenotypes (r ≥ 0.7, p-value < 0.05). Among these volatiles, the brown module was significantly correlated with (Z)-3-hexenol and (E)-3-hexen-1-ol acetate (r = 0.825, 0.755); the blue module was significantly correlated with β-caryophyllene (r = 0.81); the pink module was significantly correlated with linalool (r = 0.793). The results indicated that these modules play an important role in aroma biosynthesis in fresh leaves of tea plants.
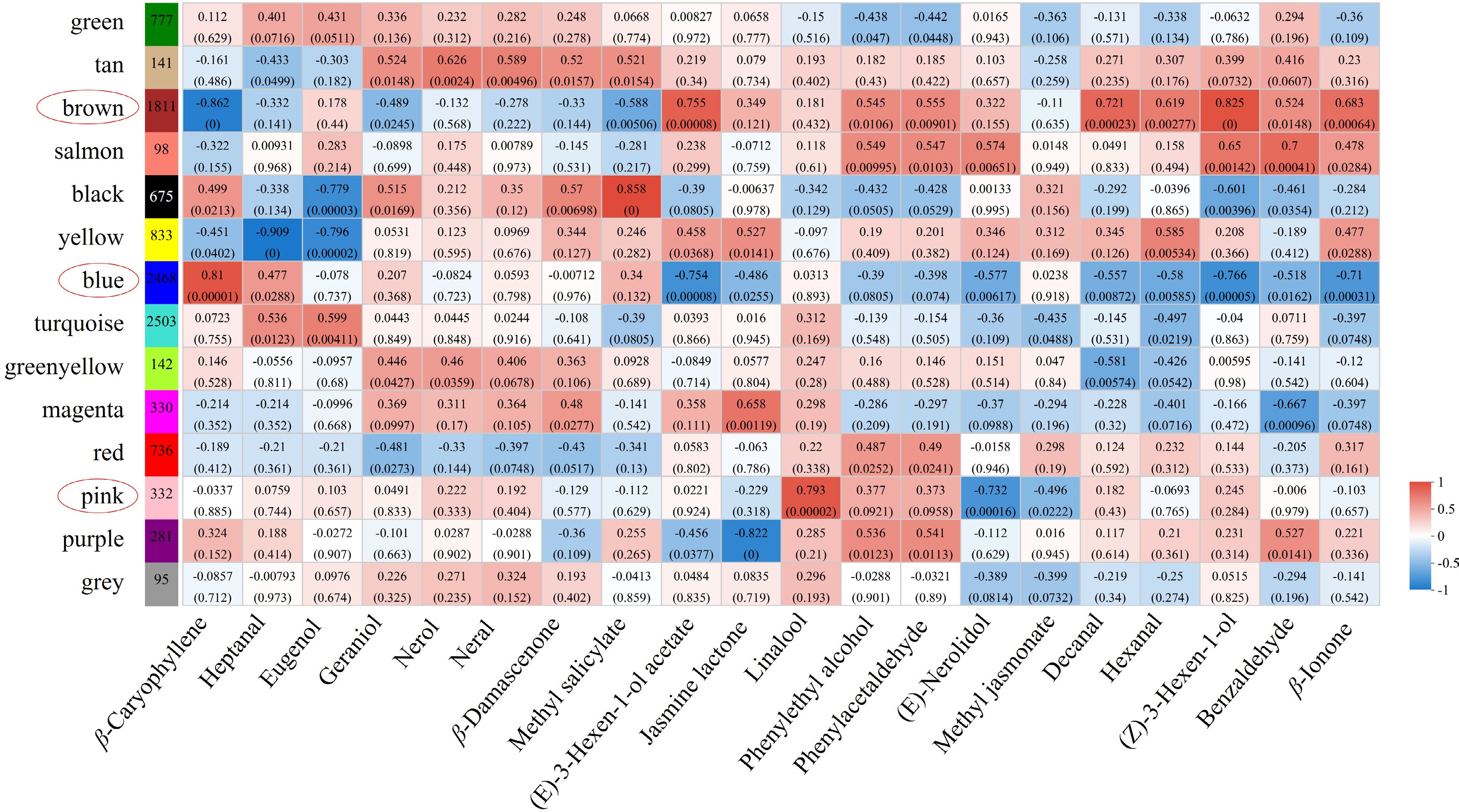
Figure 6.
Coexpression network related to key aroma compound formation. Matrix of module-metabolite associations. The abscissa represents different phenotypes, and the ordinate represents different modules. The number of genes per module is shown in the left box. Correlation coefficients and p-values between modules and metabolites are shown at the row-column intersection. Red means the module has a greater correlation with the phenotype, and blue means the module has a lower correlation with the phenotype.
To reveal the complex transcriptional regulatory network of aroma formation, genes from four modules were analysed, resulting in the identification of CsHMGR (CsTGY04G0000045), CsDXS (CsTGY02G0002953), and CsTPS (CsTGY05G0001285) genes in the blue module, two CsLIS/NES genes (CsTGY08G0001704, CsTGY08G0000359) in the pink module, and CsMVK (CsTGY05G0001238) and CsADH (CsTGY09G0001879) genes in the brown module. When r > 0.8, there may be a strong relationship between the two nodes. Therefore, transcription factors (TFs) were screened from blue, pink and brown modules. Transcriptional regulatory networks were constructed with the above genes (Fig. 7).
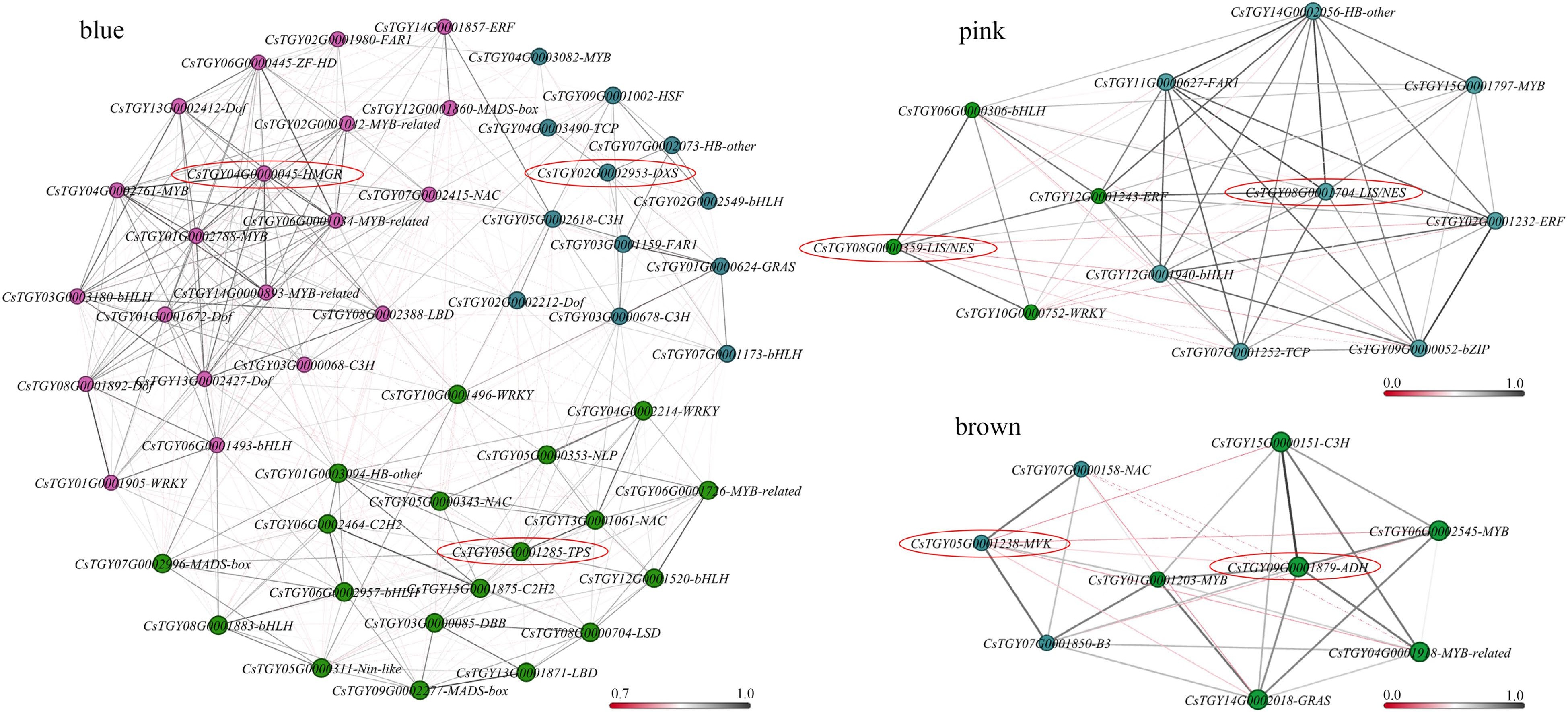
Figure 7.
Coexpression network diagram of candidate genes. When the correlation coefficient (r) is greater than 0.8, we believe that there is a regulatory relationship between the candidate genes and TFs. Nodes with the same colour indicate that the correlation coefficient between candidate genes and TFs is greater than 0.8, and the line colours between nodes indicate the strength of the correlation.
In the blue module, there were 23, 9, and 19 TFs that were significantly correlated with CsHMGR, CsDXS, and CsTPS, respectively. These TFs may be directly or indirectly involved in gene expression and β-caryophyllene biosynthesis in fresh leaves. MYB (CsTGY01G0002788, CsTGY02G0001042, CsTGY04G0002761, CsTGY06G0001034, CsTGY14G0000893), bHLH (CsTGY03G0003180), and NAC (CsTGY07G0002415) showed the strongest relationship to CsHMGR (r > 0.9); TCP (CsTGY04G0003490) and HB-other (CsTGY07G0002073) showed the strongest relationship to CsDXS (r > 0.9); TCP (CsTGY04G0003490) and HB-other (CsTGY07G0002073) showed the strongest relationship to CsDXS (r > 0.9); NAC (CsTGY13G0001061), HB-other (CsTGY01G0003094), and bHLH (CsTGY12G0001520) showed the strongest relationship to CsTPS (r > 0.9).
In the pink module, three and eight TFs were significantly correlated with CsLIS/NES, respectively. Among them, bHLH (CsTGY06G0000306) and WRKY (CsTGY10G0000752) showed the strongest relationship to CsLIS/NES1 (r > 0.9); MYB (CsTGY15G0001797), ERF (CsTGY12G0001243), bHLH (CsTGY12G0001940) and bZIP (CsTGY09G0000052) showed a significant positive correlation with CsLIS/NES2 (r > 0.8).
The brown module identified seven TFs, and these TFs may be involved in the regulation of CsMVK and CsADH genes. B3 (CsTGY07G0001850) and NAC (CsTGY07G0000158) showed a significant positive correlation with CsMVK (r > 0.8); MYB (CsTGY01G0001203, CsTGY04G0001918, CsTGY06G0002545), C3H (CsTGY15G0000151) and GRAS (CsTGY14G0002018) showed a significant correlation with CsADH (r > 0.8). These results suggested that the TFs above may be involved in regulating the characteristic volatile components and their key genes in fresh leaves of tea cultivars.
Analysis of DEG expression level by PCR
-
To verify the accuracy of the transcriptome data, the transcript abundances of eight selected DEGs were analysed by qRT-PCR. In total, four DEGs in the terpene synthesis pathway, two DEGs, and two TFs in the LOX pathway were identified (Fig. 8). The relative expression of qRT-PCR was consistent with the trend of RNA-Seq, indicating that the transcriptome sequencing results could be reliable.
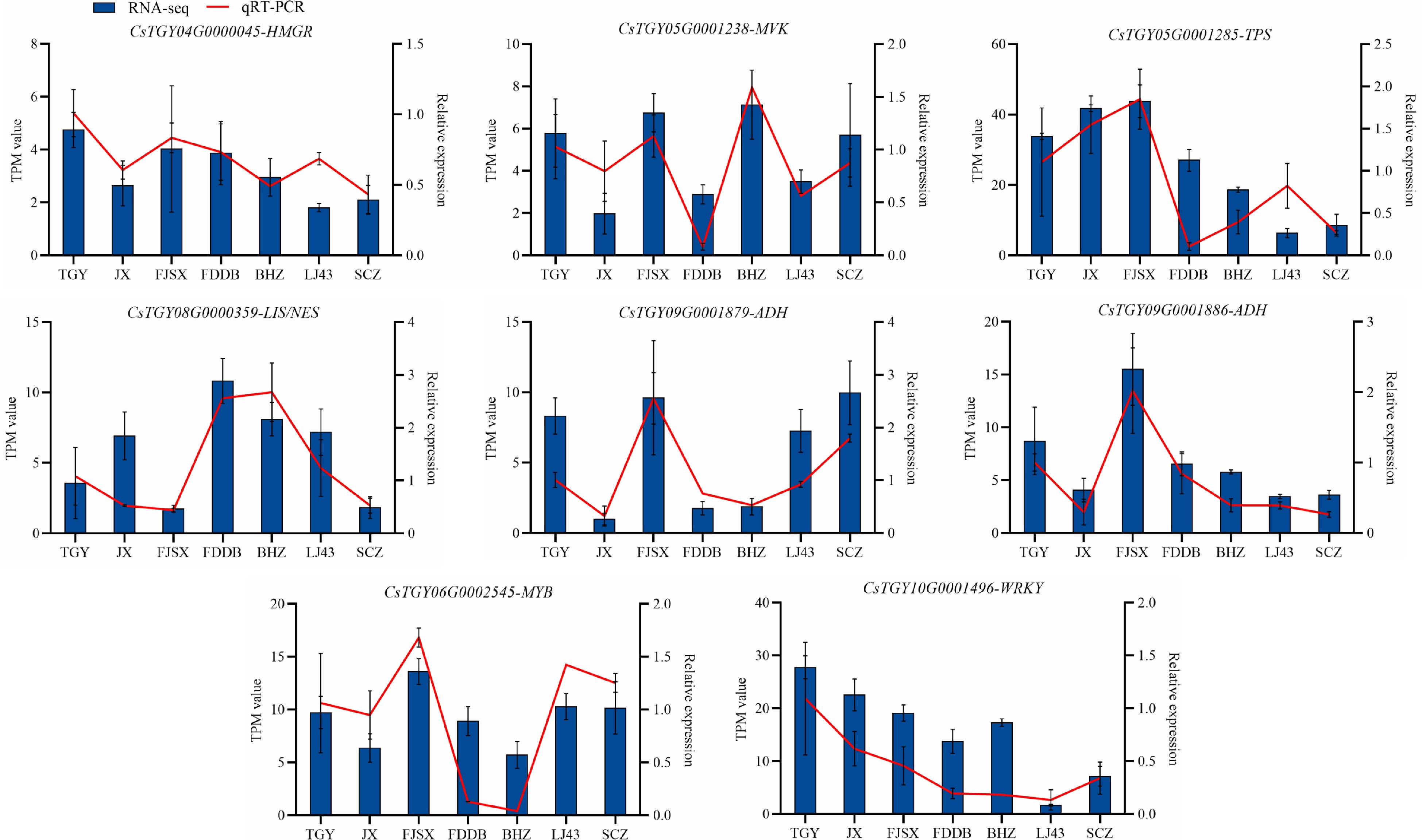
Figure 8.
Verification of the expression levels of eight differentially expressed genes.
-
Tea plant cultivars possess distinct genetic and biochemical characteristics, which largely determine their suitable tea species and quality[35]. In previous studies[18], we have found that the contents of catechin and purine alkaloids in TGY, JX, and FJSX were higher, the contents of sweet amino acids and sugars in FDDB were higher, and the contents of free amino acids and nucleotides in suitable green tea cultivars were higher. In addition to non-volatile metabolites, volatiles are also particularly important for the formation of tea quality. Aromatic substances in fresh leaves are the material basis for the formation of tea aroma[36]. Tea cultivars play an important role in the chemical compositions of fresh tea leaves and the enzymatic activities of aroma volatile-related enzymes[37]. Therefore, we further analyzed the volatile components of these seven tea cultivars to explore the reasons for their unique flavor from the raw materials of fresh leaves.
Each tea cultivar possesses its own phenotype and characteristic metabolites, some suitable for processing green tea, white, black, or oolong tea. The abundance of terpenes in fresh leaves plays an important role in the aroma quality of tea, which usually has an attractive floral and fruity aroma[38]. High ratio of terpenoid volatiles to green leaf volatiles could be regarded as a good indicator in screening cultivar for suitably producing high quality oolong tea[5]. The terpenoids in TGY and JX accounted for about 72% of the total contents, we speculated that this may be one of the reasons for their suitable preparation of oolong tea. Furthermore, the eugenol[34] and heptanal[28] may contribute to the fruity and flower characteristics of TGY, and methyl salicylate[3], (E)-β-ocimene[3], and geraniol[27] may be beneficial to JX tea aroma. During processing, Zeng et al.[39] found the GLVs and monoterpenes have relatively large changes in JX, the content of homoterpenes changed sharply in TGY. The differences in in gene expression regulation may all affect the production and concentration of terpenes in plants[40]. The content of esters in the fresh leaves of tea plants was only lower than that of alcohols, among which jasmine lactone is the key volatile component that gives oolong tea its fatty, dairy and floral characteristics[3], which may play an important role in the aroma formation of FJSX. Tea aroma intensity was significantly related to the contents of esters in tea leaves, and the higher the content, the better the quality. The triploid tea plants increase the gene dose due to the doubling of chromosomes, and the amount of transcription and expression products will inevitably change accordingly[41]. The doubling of chromosomes first leads to changes in the genomic structure, resulting in the re-regulation of gene expression and changes in gene expression levels. The results may be related to the fact that FJSX is the triploid tea resource.
Phenylethyl alcohol, phenylacetaldehyde, linalool, and its oxidized products were the main volatiles of white tea[42], we speculated that the significant accumulation of linalool and linalool oxide content in FDDB may contribute to the clear and fresh characteristics of the white tea. Aromatic alcohols with floral and fruit aromas were not abundant in the fresh leaves of tea plants[1], among which phenylethyl alcohol and phenylacetaldehyde may be the main material basis for white tea and green tea to have mellow aroma quality[6]. Green tea has a variety of flavour characteristics, such as scent types of floral, fruity, nutty, chestnut-like fragrances, and so on[43]. (Z)-3-hexenol could play a determining role in the 'raw grass' odour of finished green tea due to its overly strong and sharp green aroma[44], and (E)-3-Hexen-1-ol acetate has a grassy and fruity flavor[30]. The contents of these aroma components were higher in cultivars suitable for green tea, which may be closely related to the aroma formation.
The biosynthesis of terpenes with floral and fruit aromas is catalyzed by TPS enzymes via either the MVA or MEP pathway[45]. Our previous study[9, 30] showed that CsTPS have different functions in the synthesis of terpenoids. Studies have found that CsLIS might be involved in the regulation of linalool accumulation in the tea manufacturing process[30], and CsNES were highly expressed in oolong tea during tea turning (E)-nerolidol accumulation[46]. In this study, CsLIS/NES (CsTGY08G0001704) might be the key gene affecting the accumulation of linalool in FDDB fresh leaves, and CsNES/GIS (CsTGY08G0001826) were significantly upregulated in BHZ which might be related to the higher content of (E)-nerolidol. Volatile aliphatic components were biosynthesized from linoleic acid and linolenic acid via the lipoxygenase (LOX) pathway[37]. Studies have confirmed that COMT, CCoCOMT, CCR, and CAD are involved in the biosynthesis of eugenol[47−50]. Our results showed that the expression levels of CsCOMT (CsTGY08G0002131), CsCCoCOMT (CsTGY06G0000958), and CsCAD (CsTGY04G0002121) may be closely related to the biosynthesis of eugenol in TGY.
AP2/ERF, bHLH, WRKY, MYB, NAC, and bZIP were the common TF families that regulate terpenoid synthesis[51]. Based on WGCNA study, we speculated that bHLH, MYB, WRKY, and NAC TFs may play an important role in inducing the synthesis of β-caryophyllene by regulating CsHMGR (CsTGY04G0000045), CsDXS (CsTGY02G0002953), and CsTPS (CsTGY05G0001285). In Arabidopsis thaliana, the induction of TPS21 and TPS11 results in increased emission of sesquiterpenes, especially (E)-β-caryophyllene[52]. Overexpression of CpMYC2 and CpbHLH13 in Arabidopsis thaliana and tobacco can promote the synthesis of linalool and β-caryophyllene[53, 54]. Our study also identified that CsbHLH (CsTGY12G0001520) was annotated as MYC2, which may related to β-caryophyllene accumulation in fresh leaves by regulating the expression of the CsTPS (CsTGY05G0001285). In grape berries, VtNAC, VtC2C2-GATA, and VtbHLH were involved in the synthesis of linalool by regulating TPS genes[7]. TFs such as bHLH, WRKY, NAC, and ERF were directly involved in the regulation of linalool synthesis by binding with promoters of CsLIN[30]. Transcription factors play an essential regulatory role in the growth and development of tea plants and complex with other transcription factors to regulate plant secondary metabolism[55]. In Freesia hybrida and Arabidopsis thaliana, AtMYB21 and AtMYC2 were confirmed to participate in linalool synthesis by interacting with each other to form MYB-bHLH complex to control the expression of linalool synthase genes[8]. In this study, bHLH, WRKY, ERF, and MYB were involved in the biosynthesis of linalool in fresh leaves by regulating CsLIS/NES1 (CsTGY08G0000359) and CsLIS/NES2 (CsTGY08G0001704), especially ERF (CsTGY02G0001232) may be the key transcription factor affecting linalool synthesis. In tea plant, CsMYB is a key gene in lipid metabolism, and it mainly affects lipid metabolism by regulating CsADH to participate in aroma biosynthesis[25]. And CsMYB is involved in the biosynthesis of fatty acid derivatives by regulating the LOX pathway in green tea[56]. In the present study, we speculated that CsMYB (CsTGY01G0001203, CsTGY04G0001918, CsTGY06G0002545) may play an important role in regulating the expression of CsADH (CsTGY09G0001879) and the accumulation of (Z)-3-hexenol and (E)-3-hexene-1-ol acetate in fresh leaves.
-
In this study, we conducted a comprehensive metabolomic and transcriptomic analysis of seven tea plant cultivars to investigate the cultivar characteristic volatile components of different tea plants and their possible molecular mechanisms leading to volatile component accumulation. Overall, terpenes accounted for a large proportion of the fresh leaves of oolong tea cultivars. The aroma compositions of white and green tea cultivars were similar, and the contents of (Z)-3-hexenol, phenylethyl alcohol, phenylacetaldehyde, linalool, and its oxides were higher. The accumulation of volatile components is not only controlled by the expression of structural genes but also involved in the regulation of many transcription factors. Our study revealed the characteristic volatile components and their key regulatory genes of seven tea cultivars, which will provide a theoretical basis for breeding and suitability research of tea cultivars.
This research was funded by the Major Special Project of Scientific and Technological Innovation on Anxi Tea (Grant No. AX2021001), Fujian Agriculture and Forestry University Construction Project for Technological Innovation and Service System of Tea Industry Chain (K1520005A01).
-
The authors declare that they have no conflict of interest.
-
# These authors contributed equally: Ting Gao, Shuxian Shao
- Supplemental Table S1 The primers for qRT-PCR.
- Copyright: © 2023 by the author(s). Published by Maximum Academic Press, Fayetteville, GA. This article is an open access article distributed under Creative Commons Attribution License (CC BY 4.0), visit https://creativecommons.org/licenses/by/4.0/.
-
About this article
Cite this article
Gao T, Shao S, Hou B, Hong Y, Ren W, et al. 2023. Characteristic volatile components and transcriptional regulation of seven major tea cultivars (Camellia sinensis) in China. Beverage Plant Research 3:17 doi: 10.48130/BPR-2023-0017 

Characteristic volatile components and transcriptional regulation of seven major tea cultivars (Camellia sinensis) in China
- Received: 12 April 2023
- Revised: 14 June 2023
- Accepted: 19 June 2023
- Published online: 18 July 2023
Abstract: The volatiles in the young shoots of tea cultivars are the important material basis for the formation of tea aroma, but the cultivar-specific aroma and its molecular regulation are still lacking in research. In this study, the characteristic volatiles of seven tea cultivars in China were detected, and the results showed that the green tea cultivars 'Fuding Dabaicha' (FDDB), 'Longjing43' (LJ43), 'Shuchazao' (SCZ), and 'Baihaozao' (BHZ) were rich in (E)-3-hexenol, phenylethyl alcohol, phenylacetaldehyde, and β-ionone. For oolong tea cultivars, the characteristic volatiles of 'Tieguanyin' (TGY) were heptanal and eugenol, while the contents of (E)-β-ocimene, geraniol, and methyl salicylate were significantly increased in 'Jinxuan' (JX). In addition, 'Fujian Shuixian' (FJSX) has the highest content of esters, mainly jasmonolactone and dihydrojasmonolactone. Transcriptomic analysis showed that the different tea cultivars were significantly enriched in different levels of gene transcription in the three pathways related to aroma biosynthesis. Potential regulatory modules and genes of several characteristic volatiles were identified by WGCNA, among which CsbHLH (CsTGY12G0001520) may regulate the expression of CsTPS (CsTGY05G0001285) to directly affect the accumulation of β-caryophyllene in young shoots, while CsMYB (CsTGY01G0001203, CsTGY04G0001918, CsTGY06G0002545) may affect the synthesis of (Z)-3-hexenol and (E)-3-hexen-1-ol acetate by regulating the CsADH (CsTGY09G0001879). In addition, the transcription factors bHLH, WRKY, ERF, and MYB may be involved in the biosynthesis of linalool by regulating the expression of CsLIS/NES (CsTGY08G0001704, CsTGY08G0000359) genes individually or through interaction. These results revealed the characteristic volatiles and their key regulatory genes of seven tea cultivars, which will provide a theoretical basis for breeding and suitability research of tea cultivars.
-
Key words:
- Tea cultivars /
- Volatile metabolomics /
- Characteristic volatile components /
- Transcriptome /
- WGCNA